Breaking Therapeutic Barriers: CRISPR Screens Identify Core Targets, GSK3β Inhibitors Strike at B-ALL Survival


Introduction
Aberrant regulation of the canonical Wnt/β-catenin pathway drives tumorigenesis in multiple cancer types. However, its role in B cell acute lymphoblastic leukemia (B-ALL) has remained controversial. In a recent study published in Nature Cancer, the Cosgun laboratory and collaborators used CRISPR-based functional genomics to demonstrate that B-ALL cells rely on efficient β-catenin degradation for survival.GSK3β inhibitors block β-catenin degradation, leading to the formation of β-catenin-Ikaros/NuRD complexes, suppression of MYC transcription, and subsequent tumor cell death. This study provides a mechanistic rationale for targeting β-catenin degradation in resistant B cell malignancies and highlights GSK3β inhibition as a promising therapeutic strategy.

Research Background
Acute lymphoblastic leukemia (ALL) is a common hematologic malignancy, and conventional therapies are insufficient for a subset of patients, highlighting the urgent need for novel therapeutic targets and strategies. In the canonical Wnt/β-catenin pathway, aberrant degradation of β-catenin often drives tumorigenesis; however, its mechanistic role in B cell acute lymphoblastic leukemia (B-ALL) remains unclear. Glycogen synthase kinase 3β (GSK3β) is a critical kinase mediating β-catenin degradation, and GSK3β inhibitors have demonstrated favorable safety profiles in clinical studies, providing a foundation for exploring their therapeutic potential in B-ALL.
Research Objectives
- To elucidate the unique role and molecular mechanism of β-catenin degradation in B-ALL.
- To evaluate the therapeutic effect of targeting β-catenin degradation with GSK3β inhibitors.
- To provide new therapeutic avenues for refractory B cell malignancies.
Research Methods
-
Experimental Models:
Murine and human B-ALL cell lines.
Patient-derived xenograft (PDX) models.
Genetically engineered mouse models (e.g., Apc fl/+ , Gsk3b fl/+ , β-catenin S33,S45+/fl ). -
Gene Editing:
CRISPR-Cas9–mediated knockout or mutant cell lines and mice, including IKZF1/3 double knockout and β-catenin phosphorylation site deletion models. -
CRISPR Library Selection and Transduction:
Mouse genome-wide EKO sgRNA library targeting 22,956 genes with 278,754 sgRNAs (12 per gene plus negative controls). Lentiviral transduction of the library into inducible Cas9 NALM6 cells, ensuring each cell randomly carries a single sgRNA. -
Protein and Molecular Mechanism Validation:
Protein interactions assessed by immunoprecipitation, mass spectrometry, and Western blot. Gene expression and pathway changes analyzed via RNA-seq and GSEA. Chromatin binding assessed with ChIP-seq. -
Functional Validation:
Cell proliferation and death measured by flow cytometry, colony formation, and viability assays. In vivo therapeutic studies conducted in animal models to monitor tumor burden and survival. -
Drug Sensitivity Analysis:
Evaluated GSK3β inhibitor selectivity and therapeutic effect using public datasets (DepMap) and preclinical experiments.
Research Workflow
-
Phenotype Validation:
Characterize β-catenin expression and degradation dependency in B-ALL. Compare B-ALL with solid tumors and other B cell malignancies. -
Mechanistic Elucidation:
Confirm the necessity of GSK3β-mediated β-catenin degradation for B-ALL survival. Elucidate the molecular mechanism by which β-catenin forms a complex with Ikaros/NuRD to repress MYC transcription. -
Therapeutic Validation:
Assess single-agent efficacy and selectivity of GSK3β inhibitors in cell lines, organoids, and animal models. -
Clinical Translation:
Validate the therapeutic potential of GSK3β inhibitors in refractory B-ALL using clinical samples and PDX models.
Key Findings
-
β-Catenin Is Lowly Expressed and Dependent on Efficient Degradation in B-ALL
Compared with solid tumors, β-catenin mRNA levels in murine and human B-ALL cells are largely normal, yet protein abundance is extremely low—approximately 1/32 of that observed in solid tumors. In B-ALL cells, the N-terminal region of β-catenin is continuously phosphorylated by GSK3β, maintaining it in a state of persistent proteasomal degradation. No stable, non-phosphorylated, transcriptionally active form of β-catenin was detected.Analysis of clinical samples revealed that pathogenic alterations in β-catenin degradation pathway components—including CTNNB1, APC, and AXIN1—occur at significantly lower frequencies in B cell malignancies compared with solid tumors, highlighting a unique reliance on efficient β-catenin turnover for B-ALL cell survival.
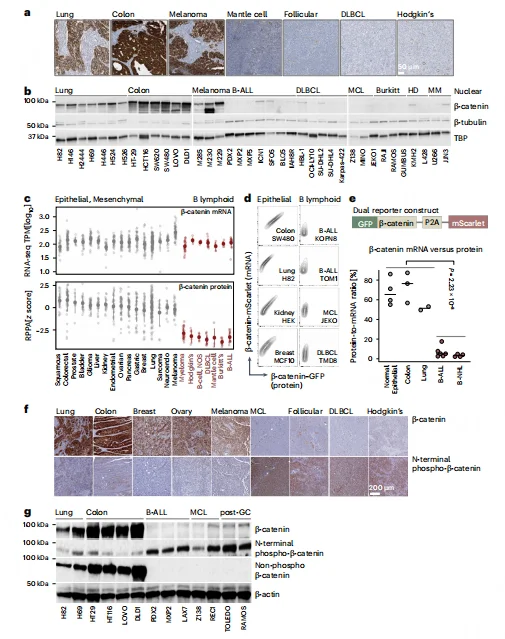
Figure 1. B Lymphocytes Express β-Catenin mRNA but Lack β-Catenin Protein
-
β-Catenin Degradation Is Essential for B-ALL Survival and Leukemia Initiation
Genetic knockout experiments demonstrated that monoallelic loss of APC, GSK3β, or CK1α leads to accumulation of β-catenin, which induces acute cell death and abolishes colony-forming capacity in B-ALL cells. In vivo, blocking β-catenin degradation significantly reduced leukemia-initiating cell (LIC) frequency from 1/1,042 to 1/40,063 and prolonged survival of transplanted mice.B cell–specific deletion of β-catenin phosphorylation sites caused developmental arrest at the pro-B cell stage, confirming that B cell maturation critically depends on efficient β-catenin degradation. Furthermore, reprogramming B-ALL cells into myeloid-like cells via CEBPα abolished their sensitivity to β-catenin accumulation, indicating that dependence on degradation is a B cell–intrinsic property.
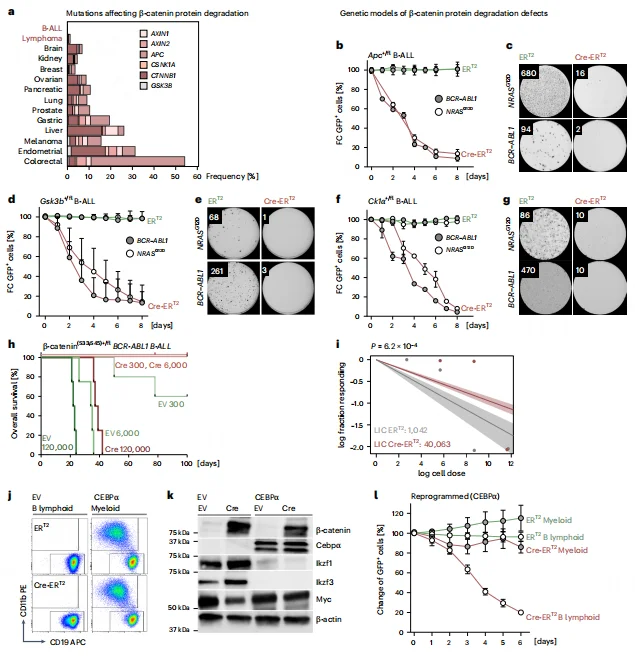
Figure 2. B Cell Malignancies Exhibit a Unique Dependence on Efficient β-Catenin Degradation
-
β-Catenin Accumulation Induces B-ALL Cell Death via MYC Suppression
RNA-seq and GSEA analyses revealed that β-catenin accumulation significantly suppresses MYC target gene sets while upregulating negative regulators of the Wnt pathway, including Axin2 and Nkd1. Dual-reporter assays demonstrated a negative correlation between β-catenin accumulation and MYC expression following GSK3β inhibitor treatment, with MYC signaling markedly downregulated after 10 hours.Functional rescue experiments showed that MYC overexpression could restore cell viability and colony-forming capacity in β-catenin–accumulated B-ALL cells. Conversely, β-catenin knockout reversed the inhibitory effect of GSK3β inhibition on MYC, confirming that β-catenin–mediated MYC suppression is the key mechanism driving B-ALL cell death.
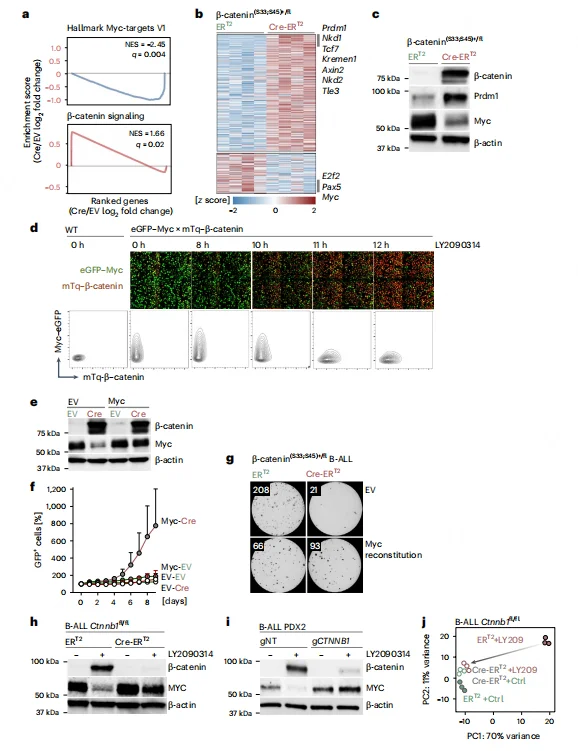
Figure 3. Impaired β-Catenin Degradation in B Cells Leads to MYC Transcriptional Suppression
-
β-Catenin Forms a Repressive Complex with Ikaros/NuRD in B Cells
Immunoprecipitation and mass spectrometry analyses revealed that in B-ALL, β-catenin does not interact with canonical TCF transcription factors. Instead, it forms complexes with B cell–specific transcription factors IKZF1/3 (Ikaros/Aiolos) and the NuRD chromatin remodeling complex (including Chd4, Mta1/2, etc.). Double knockout of IKZF1/3 disrupted the interaction between β-catenin and the NuRD complex, reversing MYC suppression and rescuing cell death, whereas single knockout of either IKZF1 or IKZF3 had no effect.Lineage comparison studies demonstrated that the β-catenin–Ikaros/NuRD interaction is B cell–specific, as β-catenin in solid tumors predominantly interacts with TCF family transcription factors. This finding highlights a lineage-restricted mechanism by which β-catenin accumulation induces transcriptional repression and cytotoxicity in B-ALL.
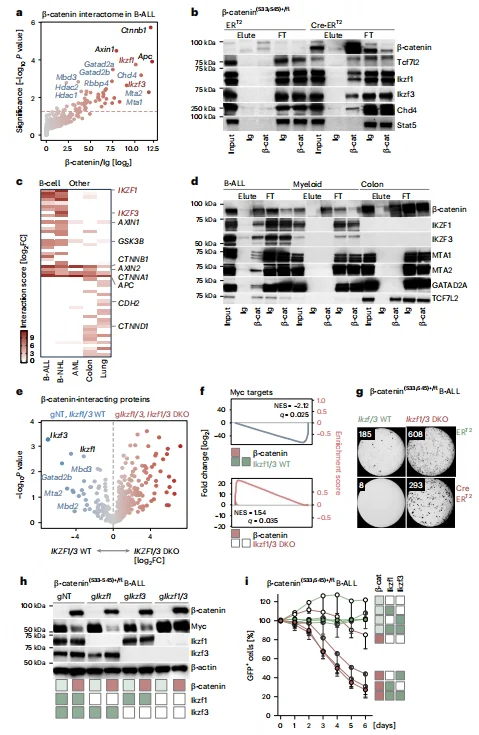
Figure 4. β-Catenin Forms a Repressive Complex with B Lineage Transcription Factors IKZF1 and IKZF3
-
Ikaros–β-Catenin Complex Mediates MYC Suppression via the BENC Enhancer
ChIP-seq analysis revealed that approximately 72.2% of β-catenin binding peaks overlap with Ikaros, with both factors co-occupying the hematopoietic-specific BENC enhancer region of MYC. Accumulation of β-catenin recruits the NuRD chromatin remodeling complex, leading to reduced H3K27ac levels at the BENC enhancer—a mark of active enhancers—thereby suppressing MYC transcription.Mutation of the IKZF core binding motif (GGGAA) within the BENC enhancer blocked β-catenin–mediated MYC repression, maintaining cell survival and competitive fitness. These findings establish a B cell–specific epigenetic mechanism in which β-catenin accumulation directly links transcription factor engagement to enhancer remodeling and MYC downregulation.
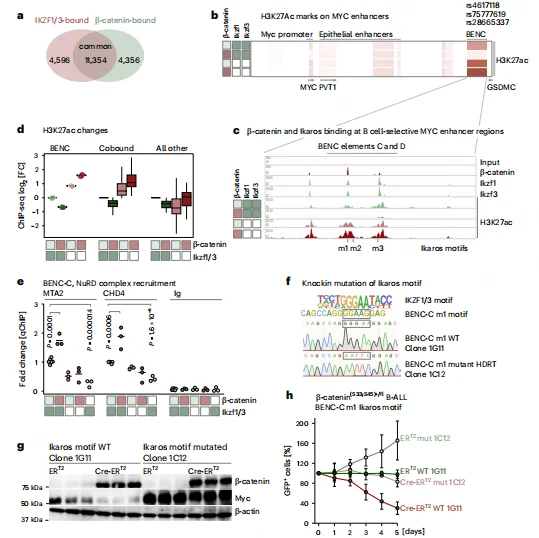
Figure 5. Identification of the Ikaros Motif in the MYC BENC Enhancer Is Required for β-Catenin–Mediated MYC Suppression
-
GSK3β Inhibitors Exhibit Selective Therapeutic Activity in B-ALL
Drug sensitivity analysis revealed that GSK3β inhibitors (e.g., LY2090314, CHIR99021) exhibit significantly higher cytotoxicity in B cell malignancies compared with solid tumors, with B-ALL EC50 values approximately 176-fold lower than those of solid tumor lines.CRISPR-based chemical-genetic screens confirmed that CTNNB1 (β-catenin) knockout most robustly rescues B-ALL cells from GSK3β inhibitor–induced cell death, validating β-catenin degradation as the central therapeutic target. B-ALL cells lacking β-catenin (e.g., BV173, PDX2) were completely resistant to GSK3β inhibitors, further demonstrating that drug efficacy depends on β-catenin accumulation. Gene expression correlation analysis indicated that high IKZF1/3 expression is positively associated with GSK3β inhibitor sensitivity, whereas high TCF7L1/2 expression correlates with resistance, highlighting lineage-specific determinants of therapeutic response.
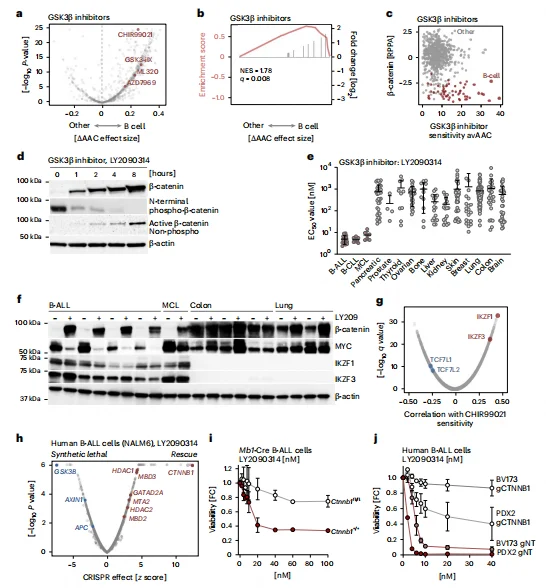
Figure 6. Small-Molecule Inhibition of β-Catenin Degradation Induces B Cell–Selective Death
-
Clinical Translation Potential of GSK3β Inhibitors in Refractory B Cell Malignancies
GSK3β inhibitors were effective in both chemotherapy-sensitive and refractory B-ALL PDX models, independent of IKZF1 deletion status. B cell lymphomas harboring MYC translocations (8q24+) exhibited markedly reduced sensitivity to GSK3β inhibitors, providing a basis for patient stratification in clinical settings.In vivo, single-agent LY2090314 treatment significantly reduced tumor burden in refractory B-ALL and mantle cell lymphoma (MCL) PDX models and prolonged mouse survival (P = 6.5 × 10⁻⁴ to 1.2 × 10⁻⁵). Treated mice showed no overt organ toxicity, and the phenotypic characteristics of B-ALL cells remained largely unchanged, supporting the safety and lineage-specificity of this therapeutic approach.
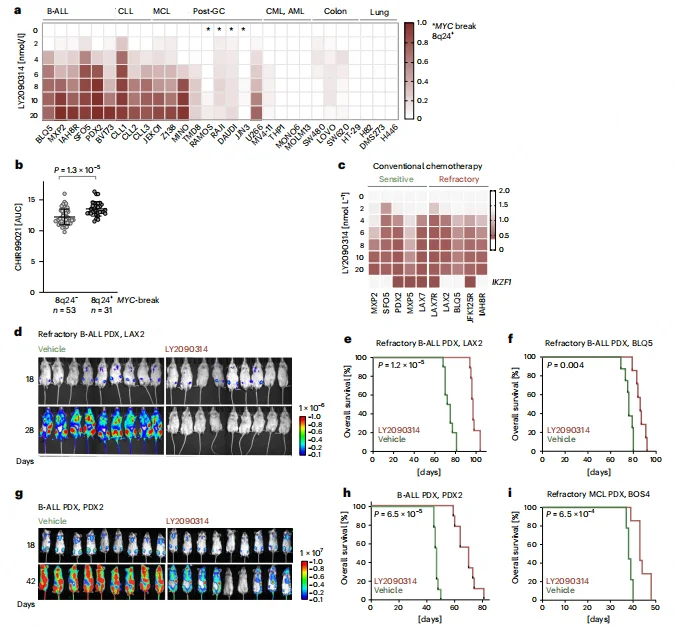
Figure 7. Rationale for Repurposing GSK3β Inhibitors in Refractory B Cell Malignancies
Summary
This study uncovers a B-ALL-specific dependency on efficient β-catenin degradation for cell survival. GSK3β-mediated β-catenin turnover prevents its interaction with the Ikaros/NuRD complex, thereby maintaining MYC expression and promoting tumor cell viability.Pharmacologic inhibition of GSK3β blocks β-catenin degradation, leading to β-catenin accumulation, suppression of MYC transcription, and B cell-specific tumor cytotoxicity.These findings reveal a noncanonical role of β-catenin in B-ALL and highlight the translational potential of clinically accessible GSK3β inhibitors as a targeted therapy for refractory B cell malignancies.
Ubigene CRISPR-iScreen™ Library
- Over 40 ready-to-use CRISPR libraries covering human, mouse, green monkey, and pig genomes, including genome-wide knockout, inhibition, and activation libraries.
- Sub-libraries targeting kinases, cell cycle regulators, membrane proteins, metabolism-related genes, and more for focused functional screens.
- 50+ CRISPR library lentivirus and 400+ CRISPR Library cell pools available for immediate use.
- One-stop CRISPR functional screening services for in vitro and in vivo studies, supporting end-to-end research needs.
Free access to iScreenAnlys™ platform, with Drug-Z/MAGeCK-MLE/MAGeCK-RRA for one-click screening data analysis.Accelerate your target discovery and functional genomics research with Ubigene CRISPR-iScreen™.
Contact us to learn more>>>Reference
Cosgun KN, Jumaa H, Robinson ME, Cheng Z, Oulghazi S, Kume K, Fonseca Arce D, Agadzhanian N, Kistner KM, Leveille E, Drivet E, Yu F, Qian Z, Song JY, Chan WC, Xu L, Xiao G, Taketo MM, Kothari S, Davids MS, Schjerven H, Jellusova J, Müschen M. Targeting β-catenin degradation with GSK3β inhibitors induces cell death in acute lymphoblastic leukemia. Nat Cancer. 2026 Jan 8. doi: 10.1038/s43018-025-01093-z. Epub ahead of print. PMID: 41507538.
 Promotions
Promotions 








