Research Frontier | Uridine Emerges as a Potential Anticancer “Game Changer”?


Introduction
Immune checkpoint blockade (ICB) therapy has revolutionized cancer treatment by delivering unprecedented clinical benefits. However, its widespread application remains severely constrained by high costs and limited response rates, with only 10-30% of patients deriving durable benefit. Accumulating evidence indicates that metabolic dysregulation within the tumor microenvironment (TME) is a central driver of CD8⁺ T cell dysfunction and resistance to ICB therapy.
In a recent study published in Cell Metabolism (Impact Factor: 30.9), a research team from Southern Medical University reports a previously unrecognized mechanism by which Sorting Nexin 17 (SNX17) remodels the tumor immune microenvironment through regulation of uridine metabolism. Specifically, SNX17 stabilizes the transcription factor RUNX2, thereby upregulating uridine phosphorylase 1 (UPP1) expression and accelerating uridine catabolism within the TME. This depletion of uridine suppresses CD45 N-glycosylation and subsequent activation of the LCK-ZAP70 signaling cascade, ultimately impairing CD8⁺ T cell-mediated antitumor immunity. Notably, exogenous uridine supplementation effectively restores CD8⁺ T cell activation and function, overcoming ICB resistance in tumors with high SNX17 expression. These findings identify uridine metabolism as a critical immunoregulatory axis in the TME and highlight uridine supplementation as a promising, low-cost strategy to enhance immunotherapy efficacy in resistant tumors.

Research Background
CD8⁺ T cells constitute the central effector population in tumor immune surveillance, and their functionality is tightly regulated by the metabolic landscape of the tumor microenvironment (TME). Protein glycosylation, a critical post-translational modification, is essential for T cell development, activation, and signal transduction. However, how tumor-associated metabolic reprogramming influences T cell N-glycosylation and thereby modulates antitumor immunity remains largely unclear.
Uridine, a key nucleoside metabolite, plays indispensable roles in nucleic acid synthesis, energy metabolism, and glycoprotein biosynthesis. Despite its fundamental metabolic importance, the immunoregulatory functions of uridine and the underlying molecular mechanisms have not been fully elucidated. Sorting Nexin 17 (SNX17), a member of the sorting nexin family, is primarily involved in endocytosis and endosomal recycling of membrane proteins. To date, the role of SNX17 in tumor immunometabolism has not been reported, positioning it as a previously unexplored and potentially critical regulatory factor.
Research Objectives
This study aims to identify key regulators underlying resistance to immune checkpoint blockade (ICB) therapy, elucidate the molecular mechanisms by which these regulators modulate TME metabolism and CD8⁺ T cell function, and evaluate the therapeutic efficacy of uridine supplementation as a low-cost immunotherapeutic strategy, both as monotherapy and in combination with ICB.
Research Methods
-
Clinical Samples and Database Analysis:
Gene expression data from the GEO database (including GSE189469 and GSE215011) and the TCGA colorectal cancer (CRC) cohort were integrated with clinical data from 44 CRC patient samples. Correlations between SNX17 and UPP1 expression levels, ICB treatment response, and patient survival outcomes were systematically analyzed. -
Animal Models:
Tumor cell lines with SNX17, UPP1, or RUNX2 knockout (KO) or overexpression (OE) were generated and used to establish subcutaneous and orthotopic tumor models, including CMT93, MC38, Hepa1-6, and B16F10. Experiments were conducted in C57BL/6 mice, BALB/c nude mice, and genetically engineered mouse models, including SLC29A1-KO and CD45-KO mice, to assess tumor growth dynamics, immune cell infiltration, and therapeutic responses. -
Cellular Assays:
Gene knockout cell lines were generated using CRISPR-Cas9 technology. CD8⁺ T cells were isolated from mouse spleens and from human peripheral blood mononuclear cells (PBMCs). Co-culture systems, Transwell assays, flow cytometry, and Western blot analyses were employed to evaluate the effects of uridine on CD8⁺ T cell activation, cytokine production, and intracellular signaling pathways. -
Mechanistic Studies:
Metabolite levels were quantified using mass spectrometry-based metabolomics. Protein-protein interactions and glycosylation modifications were characterized using immunoprecipitation (IP), chromatin immunoprecipitation (ChIP), and MAL II lectin staining. Fluorescent reporter assays were applied to delineate regulatory relationships within the signaling pathways of interest. -
Therapeutic Validation:
The antitumor efficacy of exogenous uridine administration—via intraperitoneal injection or dietary supplementation—was evaluated in tumor-bearing mice, both as a single agent and in combination with anti-PD-1 antibody therapy. Functional remodeling of immune cell populations within the TME was comprehensively assessed.
Study Workflow
-
Resistance Factor Identification:
Bioinformatic analyses were performed to identify SNX17 as a key determinant of resistance to immune checkpoint blockade (ICB). Its association with CD8⁺ T cell infiltration and patient prognosis was systematically validated across multiple datasets. -
Functional Validation:
Using in vitro and in vivo models, we demonstrated that SNX17 suppresses CD8⁺ T cell–mediated antitumor immunity by depleting uridine within the tumor microenvironment (TME). Importantly, exogenous uridine supplementation effectively reversed this immunosuppressive phenotype. -
Mechanistic Elucidation:
We delineated the SNX17–RUNX2–UPP1 regulatory axis as a central pathway governing uridine metabolism in tumors. Mechanistically, uridine was shown to enhance CD45 N-glycosylation, thereby activating the LCK–ZAP70 signaling cascade. The functional indispensability of the CD45 N-glycosylation site N380 was further validated. -
Therapeutic Evaluation:
The therapeutic efficacy of uridine supplementation, alone or in combination with anti–PD-1 therapy, was assessed, with particular emphasis on its superior antitumor activity in tumors exhibiting high SNX17 expression.
Key Findings
-
High SNX17 Expression Is Associated with Poor Immunotherapy Response and Suppressed Antitumor Immunity
Comprehensive bioinformatic analyses identified SNX17 as the sole ICB resistance–associated candidate gene exhibiting a significant negative correlation with CD8⁺ T cell infiltration in colorectal cancer (CRC). Tumors with high SNX17 expression displayed markedly reduced proportions of CD8⁺ T cells. Consistent with these findings, SNX17 expression was significantly elevated in CRC tumor tissues compared with adjacent normal tissues. Patients with high SNX17 expression exhibited significantly shorter overall survival. Moreover, SNX17 levels were substantially higher in patients with progressive disease (PD) than in those achieving partial response or stable disease (PR + SD) following ICB therapy.In vivo studies further corroborated the immunosuppressive role of SNX17. CMT93 tumors overexpressing SNX17 were refractory to PD-1 blockade, whereas SNX17 deletion markedly enhanced tumor sensitivity to ICB therapy. Notably, SNX17 knockout did not affect tumor cell proliferation in vitro or tumor growth in immunodeficient nude mice. In contrast, SNX17 deficiency significantly restrained tumor progression in immunocompetent C57BL/6 mice, indicating a strictly immune-dependent mechanism. This antitumor effect was consistently observed across multiple tumor models, including MC38, Hepa1-6, and B16F10.
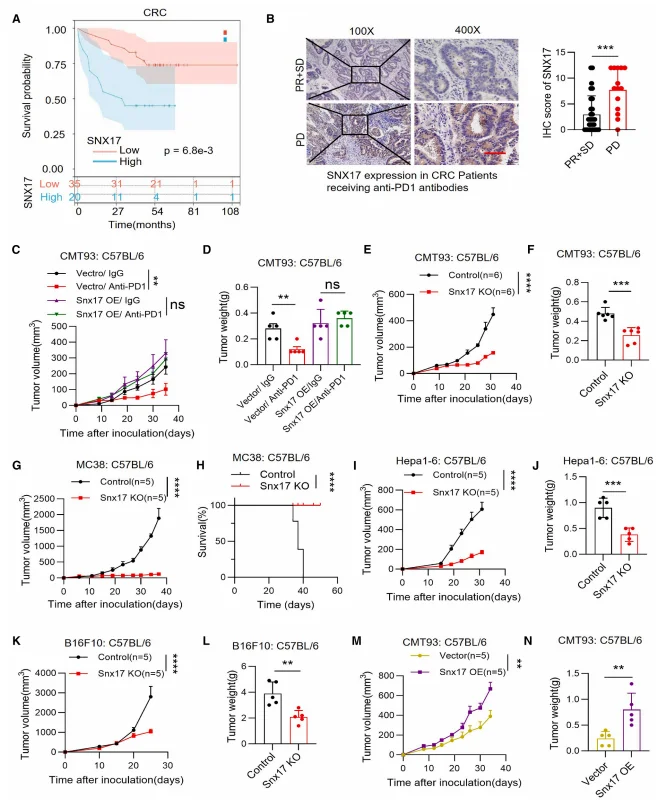
Figure 1. Elevated SNX17 Expression in Tumors Is Associated with Poor Response to Immunotherapy and Suppressed Antitumor Immunity
-
SNX17 Restricts Antitumor Immunity in a CD8⁺ T Cell–Dependent Manner
Single-cell RNA sequencing revealed a marked increase in the proportions of CD4⁺ and CD8⁺ T cells in SNX17-deficient tumors, accompanied by significant enrichment of T cell activation–related pathways. Effector molecules, including interferon-γ (IFN-γ), granzyme B (GZMB), and interleukin-2 (IL-2), were substantially upregulated, whereas the expression of T cell exhaustion markers—such as PD-1 (PDCD1), HAVCR2 (TIM-3), and TIGIT—was notably reduced.In vivo antibody-mediated depletion experiments further demonstrated that depletion of CD8⁺ T cells completely abrogated the antitumor effects conferred by SNX17 knockout, whereas depletion of CD4⁺ T cells had no significant impact. These findings establish that SNX17 mediates immunosuppression predominantly through targeting CD8⁺ T cell–dependent antitumor immune responses.
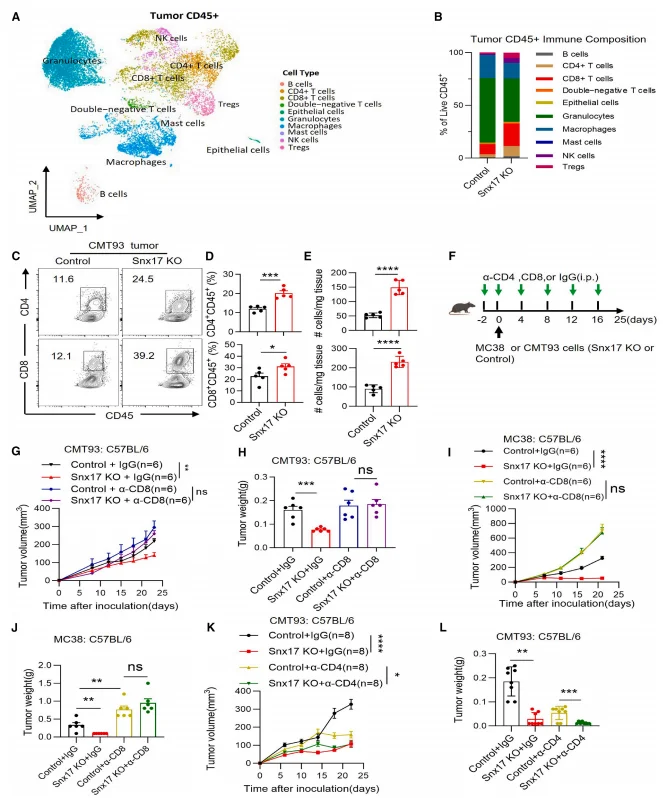
Figure 2. SNX17 Restricts CD8⁺ T Cell–Dependent Antitumor Immunity
-
SNX17 Promotes Uridine Consumption via UPP1, Thereby Suppressing CD8⁺ T Cell Activity
Mass spectrometry–based metabolomic analyses revealed a pronounced accumulation of uridine accompanied by a reduction in uracil levels in the supernatants of SNX17-deficient tumor cells as well as in SNX17 knockout tumor tissues. Conversely, enforced SNX17 overexpression significantly reduced extracellular and intratumoral uridine concentrations, indicating that SNX17 accelerates uridine catabolism.Co-culture experiments further demonstrated a competitive relationship for uridine between tumor cells and CD8⁺ T cells. Under conditions of limited uridine availability, an increased proportion of tumor cells markedly impaired uridine uptake by CD8⁺ T cells, resulting in diminished T cell activity. Functionally, exogenous uridine supplementation dose-dependently enhanced cytokine production—including interferon-γ (IFN-γ), interleukin-2 (IL-2), and tumor necrosis factor-α (TNF-α)—in both murine and human CD8⁺ T cells. This immunostimulatory effect was strictly dependent on the uridine transporter SLC29A1, underscoring the requirement for uridine uptake in sustaining CD8⁺ T cell effector function.
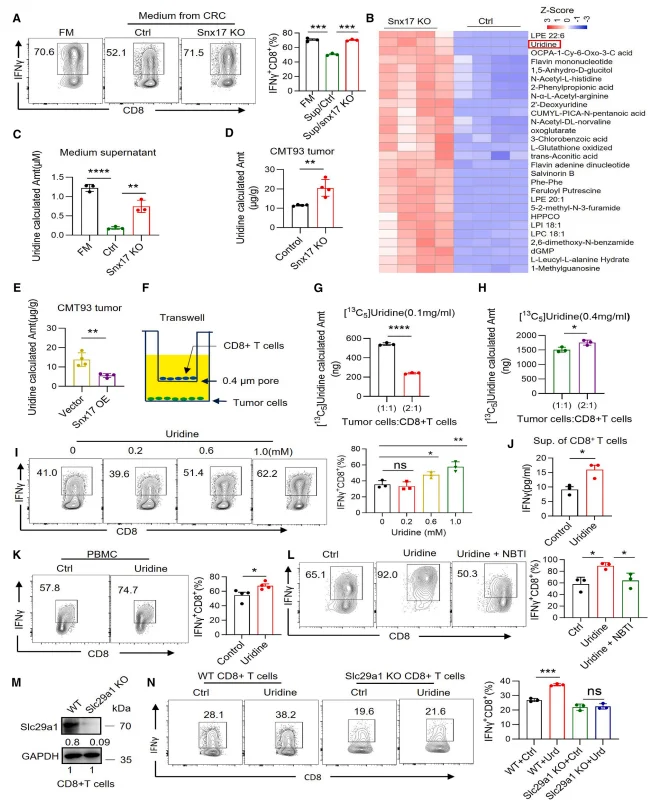
Figure 3. SNX17 Regulates CD8⁺ T Cell Activity by Promoting Uridine Consumption
-
Uridine Potentiates CD8⁺ T Cell–Mediated Antitumor Immunity
Both intraperitoneal administration and dietary supplementation of uridine significantly suppressed tumor growth in immunocompetent mice across subcutaneous and orthotopic tumor models. This antitumor effect was strictly dependent on CD8⁺ T cells, as depletion of CD8⁺ T cells abolished the therapeutic benefit of uridine supplementation.As a monotherapy, uridine exhibited antitumor efficacy comparable to that of anti–PD-1 or anti–PD-L1 antibody treatment in tumors with low SNX17 expression. Notably, in tumors characterized by high SNX17 expression, uridine supplementation displayed a pronounced synergistic effect when combined with anti–PD-1 therapy, resulting in substantially enhanced tumor control.
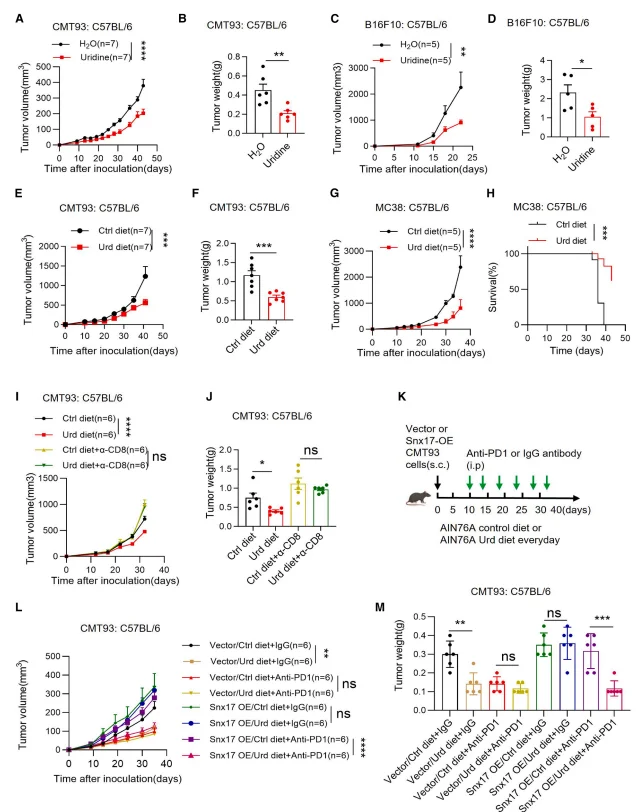
Figure 4. Uridine Enhances CD8⁺ T Cell–Mediated Antitumor Immunity
-
Uridine Activates the LCK–ZAP70 Signaling Cascade via CD45 N-Glycosylation
Levels of UDP–N-acetylglucosamine (UDP-GlcNAc), a key metabolite supporting protein N-glycosylation, were markedly increased in uridine-treated and SNX17-deficient CD8⁺ T cells. Pharmacological inhibition of N-glycosylation with tunicamycin (TM) completely abolished uridine-induced interferon-γ (IFN-γ) production, whereas inhibition of O-glycosylation had no appreciable effect, indicating a specific requirement for N-glycosylation.Mass spectrometry–based glycoproteomic analyses and MAL II lectin staining demonstrated that uridine robustly enhanced CD45 N-glycosylation. Genetic ablation of CD45 or expression of an N-glycosylation–deficient CD45 mutant (CD45RO^N380) eliminated the ability of uridine to activate CD8⁺ T cells, establishing CD45 N-glycosylation as a critical downstream effector of uridine signaling.
Mechanistically, uridine treatment promoted phosphorylation of LCK at Tyr394, as well as downstream phosphorylation of ZAP70 and CD3ζ. This phosphorylation cascade was blocked by tunicamycin, and pharmacological inhibition of LCK with PP2 reversed uridine-induced IFN-γ secretion, confirming that uridine enhances CD8⁺ T cell activation through the CD45–LCK–ZAP70 signaling axis. Consistent with these findings, UPP1 knockout phenocopied SNX17 deficiency by increasing uridine availability within the tumor microenvironment, augmenting CD8⁺ T cell IFN-γ production, and suppressing tumor growth. Conversely, UPP1 overexpression abrogated the antitumor effects observed in SNX17-deficient tumors, further validating the functional importance of the SNX17–UPP1–uridine metabolic pathway.
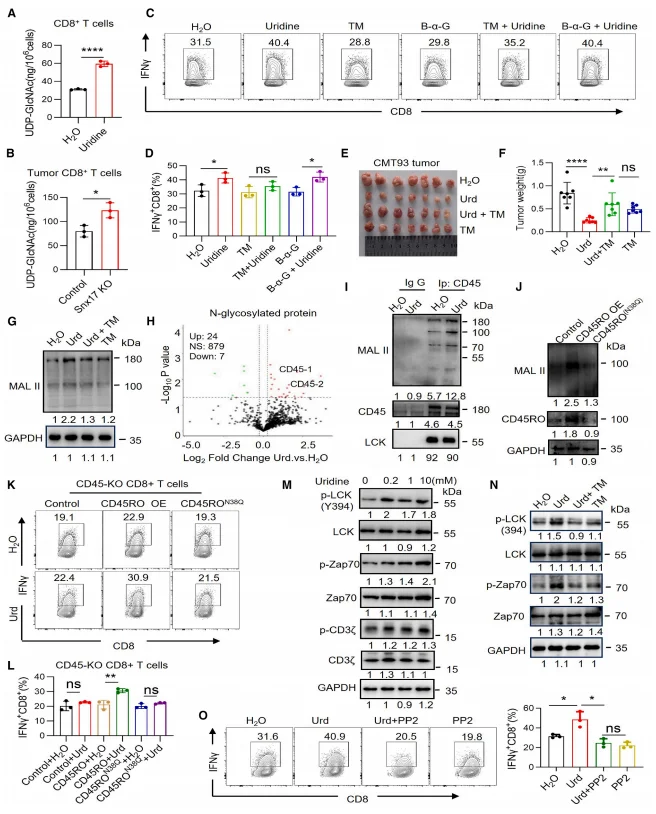
Figure 5. Uridine Enhances CD8⁺ T Cell Activity by Promoting CD45 N-Glycosylation
-
SNX17 Regulates UPP1 Transcription via RUNX2
RNA sequencing analyses, together with independent validation assays, revealed that genetic ablation of SNX17 resulted in a marked reduction in UPP1 mRNA and protein expression. Conversely, enforced overexpression of SNX17 significantly upregulated UPP1 expression at both the transcript and protein levels, indicating that SNX17 positively regulates UPP1 expression at the transcriptional level.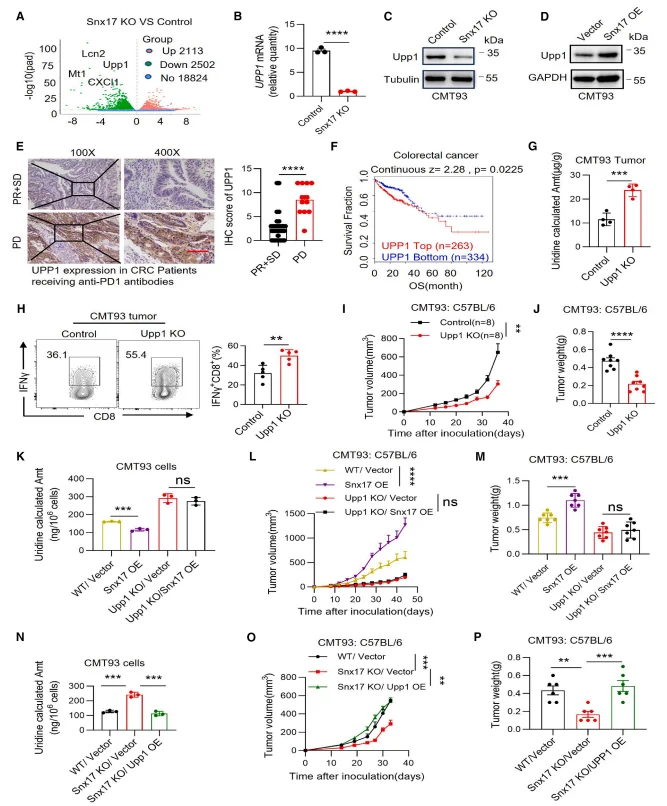
Figure 6. SNX17 Reduces Uridine Levels through UPP1
Chromatin immunoprecipitation (ChIP) assays demonstrated that RUNX2 directly binds to the promoter region of UPP1, indicating that UPP1 is a direct transcriptional target of RUNX2. Co-immunoprecipitation analyses further revealed a physical interaction between SNX17 and RUNX2. Mechanistically, SNX17 binding protected RUNX2 from lysosomal degradation, thereby enhancing RUNX2 protein stability. Functionally, genetic ablation of RUNX2 abolished the SNX17 overexpression–induced upregulation of UPP1 and reversed the tumor growth–promoting effects of SNX17 overexpression. These results establish the SNX17–RUNX2–UPP1 axis as a critical regulatory pathway controlling uridine metabolism and tumor progression.
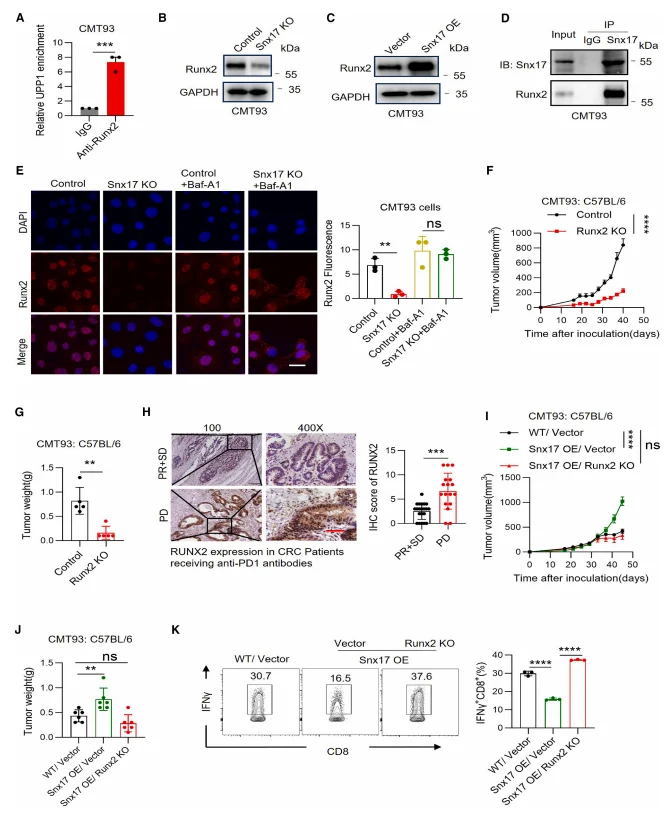
Figure 7. SNX17 Regulates UPP1 Transcription via RUNX2
Significance and Novelty
-
Mechanistic Innovation:
This study uncovers, for the first time, a SNX17–RUNX2–UPP1–uridine metabolic axis that regulates CD8⁺ T cell functionality. It defines CD45 N-glycosylation as a central mediator of uridine-induced T cell activation, expanding our understanding of how metabolites modulate immune cell function. -
Target Identification:
SNX17 and UPP1 are established as key biomarkers of ICB resistance, providing a rational basis for stratifying patients with potential responsiveness to immunotherapy. -
Therapeutic Strategy:
Exogenous uridine supplementation is proposed as a low-cost immunotherapeutic approach, particularly effective in high SNX17-expressing, ICB-resistant tumors. Uridine also synergizes with anti-PD-1 therapy, offering a feasible translational pathway for clinical application.
Summary
Through multi-layered experimental validation, this study demonstrates that SNX17 stabilizes RUNX2, thereby promoting UPP1 transcription and accelerating uridine degradation within the tumor microenvironment. This process suppresses CD45 N-glycosylation and downstream LCK-ZAP70 signaling, leading to CD8⁺ T cell exhaustion and resistance to ICB therapy. Exogenous uridine supplementation restores CD8⁺ T cell antitumor activity and shows enhanced efficacy when combined with PD-1 blockade in high SNX17 tumors. Overall, these findings reveal a novel mechanism of tumor metabolic remodeling of the immune microenvironment and provide a specific, cost-effective immunotherapeutic strategy to overcome ICB resistance.
Ubigene has always adhered to its core mission of Make genome editing easier continuously iterating its products and services. To date, Ubigene has successfully completed over 13,000 gene editing projects and offers a comprehensive portfolio of more than 11,000 cell products, including over 8,000 knockout (KO) cell lines. Leveraging proprietary technologies, Ubigene achieves 10–20× higher gene editing efficiency compared with conventional methods. Our solutions have supported over 10,000 life science laboratories, pharmaceutical companies, and CROs worldwide with high-quality gene editing services and products.
Sorting Nexin 17 (SNX17), a member of the sorting nexin protein family, primarily regulates protein endocytosis and recycling. For researchers interested in exploring SNX17 function, Ubigene provides SNX17 Knockout cell lines in widely used models, including HeLa and 293T. In addition, Ubigene offers customized gene editing solutions tailored to specific research needs. For personalized projects or complex editing strategies, our experts are ready to provide guidance and support to accelerate your research.
Contact us to learn more>>>Reference
Xiao J, Li Z, Ding Y, Zhu K, Zheng Z, Zhang Y, Weng J, Wang F, Zhang Y, Zeng S, Qiu M, Zhang Z, Wang Z, Liang L. Uridine depletion impairs CD8⁺ T cell antitumor activity through N-glycosylation. Cell Metab. 2025 Dec 29:S1550-4131(25)00530-3. doi: 10.1016/j.cmet.2025.11.016. Epub ahead of print. PMID: 41468885.
 Promotions
Promotions 








