Acetate in Cancer: Driving Tumor Growth and Immune Evasion


Introduction
Tumor cells are notorious not only for their uncontrolled proliferation, but also for their remarkable ability to evade immune surveillance. Beyond rapid growth, cancer cells actively disguise themselves to escape detection and elimination by the immune system. While glucose and glutamine metabolism have long been recognized as central drivers of tumor progression and immune modulation, an important question remains: are there other, less obvious metabolites that quietly support tumor immune evasion? A breakthrough study published in Nature Metabolism in 2024 provides a compelling answer. The study identifies acetate—a small, ubiquitous metabolite commonly derived from daily diet—as a previously unrecognized key regulator of tumor immune escape.
In this work, researchers employed A549 lung cancer cells engineered by Ubigene to carry site-specific MYC point mutations, including MYC (p.K148R) and MYC (p.K148Q). These mutations were designed to mimic the non-acetylated and acetylated states of c-Myc, respectively, enabling precise dissection of how acetylation-dependent regulation of c-Myc influences tumor cell behavior. The study uncovered a novel “metabolism-epigenetics-immunity” axis, revealing how acetate can be metabolically repurposed into a signaling molecule that enhances immune camouflage in cancer cells. This discovery reshapes our understanding of tumor metabolism and highlights acetate as a critical, yet previously underestimated, contributor to immune evasion.

Research Background
Rapid tumor growth often results in metabolic stress caused by limited nutrient availability, particularly glucose deprivation. To sustain survival and proliferation under such conditions, cancer cells actively seek alternative energy sources. Acetate, an energy precursor derived from dietary intake (e.g., fermented foods and processed meats) and gut microbiota metabolism, has recently emerged as a potential contributor to tumor metabolism.In parallel, to evade elimination by the host immune system—especially by CD8⁺ cytotoxic T lymphocytes (CTLs)—tumor cells frequently upregulate programmed death-ligand 1 (PD-L1). PD-L1 functions as a potent “don’t kill me” signal, binding to PD-1 on T cells and inducing T-cell exhaustion or functional suppression.However, whether acetate participates in the regulation of PD-L1 expression and tumor immune evasion, and the molecular mechanisms underlying such involvement, remained completely unknown prior to this study.
Research Objectives
This study systematically delineates a previously unrecognized signaling cascade:Acetate uptake via MCT1 → conversion to acetyl-CoA → DLAT-mediated acetylation of c-Myc at lysine 148 (K148) → recruitment of USP10 and stabilization of c-Myc → c-Myc-dependent transcriptional activation of PD-L1 and other target genes. The study further demonstrates that upregulated PD-L1 expression and reduced infiltration of activated CD8⁺ T cells synergistically promote tumor immune escape.
Research Methods
A comprehensive combination of multi-omics approaches and rigorous in vitro and in vivo models was employed to validate this pathway:
- Clinical Sample Analysis: Isotope tracing and metabolic flux mass spectrometry were performed on human non-small cell lung cancer (NSCLC) tissues, identifying acetate as the most abundant short-chain fatty acid within tumor samples.
- Cell and Animal Models: In cultured lung cancer cells and murine lung cancer models, isotope-labeled acetate was used to trace its metabolic fate, leading to the identification of downstream metabolic and signaling pathways.
- Molecular Mechanism Investigation: A combination of gene editing, protein–protein interaction assays, and mass spectrometry–based analyses was employed to systematically dissect the interactions among acetate, MCT1, DLAT, c-Myc, and USP10.
- Immune Function Assessment: In vitro co-culture assays and tumor microenvironment analyses were conducted to evaluate acetate-mediated suppression of T-cell function. In addition, subcutaneous tumor models were established in mice, followed by dietary acetate supplementation, treatment with MCT1 inhibitors, and combination therapy with anti–PD-1 antibodies, to assess tumor growth and immune landscape remodeling.
- Clinical Correlation Analysis: Immunohistochemical staining of NSCLC patient samples was performed to analyze correlations between c-Myc acetylation levels, MCT1 and PD-L1 expression, and CD8⁺ T-cell infiltration.
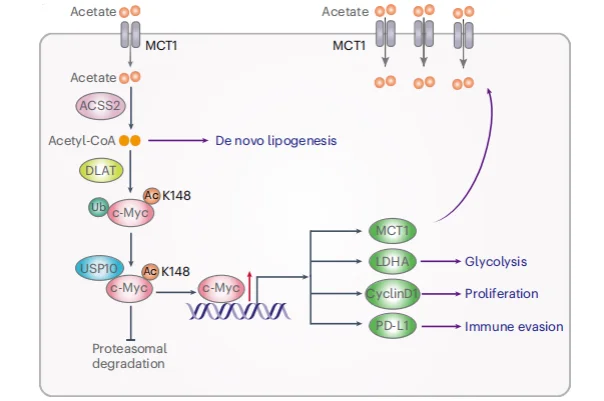
Research Workflow
- Acetate Uptake: In non–small cell lung cancer (NSCLC) tissues, acetate was identified as the most abundant short-chain fatty acid. Tumor cells exhibited elevated expression of monocarboxylate transporter 1 (MCT1), enabling efficient uptake and accumulation of acetate.
- Acetylation Modification: Intracellular acetate is converted into acetyl–coenzyme A (acetyl-CoA). The metabolic enzyme dihydrolipoamide acetyltransferase (DLAT) catalyzes acetylation of c-Myc at lysine 148 (K148).
- c-Myc Stabilization: Acetylated c-Myc recruits the deubiquitinase USP10, preventing proteasomal degradation and thereby significantly enhancing c-Myc stability and intracellular abundance.
- Transcriptional Reprogramming and Immune Modulation: In murine models, dietary acetate supplementation promoted tumor growth and suppressed CD8⁺ T-cell infiltration. Conversely, disruption of this pathway—such as inhibition of MCT1 or USP10—potentiated the therapeutic efficacy of anti–PD-1 immunotherapy.
- Clinical Relevance: In human NSCLC samples, c-Myc acetylation levels were positively correlated with MCT1 and PD-L1 expression, and negatively correlated with CD8⁺ T-cell infiltration. Elevated c-Myc acetylation was further associated with poor patient prognosis.
Key Results
-
Dietary Acetate Alleviates Energy Stress and Promotes Lung Tumor Growth in an MCT1-Dependent Manner:
Isotope tracing and metabolic flux mass spectrometry analyses demonstrated that acetate is the most abundant short-chain fatty acid in human lung cancer tissues. Tumor tissues exhibited a markedly enhanced capacity to import and enrich acetate via MCT1, converting it into acetyl-CoA at levels far exceeding those observed in normal tissues (Figure 1).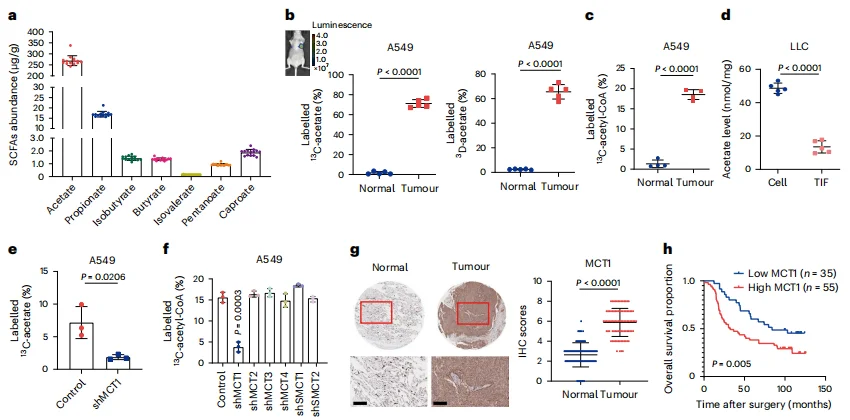
Figure 1. Tumors Depend on MCT1 for Acetate Uptake and Enrichment
In both in vitro and in vivo models, dietary supplementation of acetate significantly promoted tumor growth. Conversely, when the MCT1 transporter was inhibited or glucose was deprived—forcing cancer cells to rely on acetate—acetate supplementation rescued tumor growth (Figure 2k, m–n), demonstrating the critical dependence of tumors on acetate metabolism for proliferation under metabolic stress.
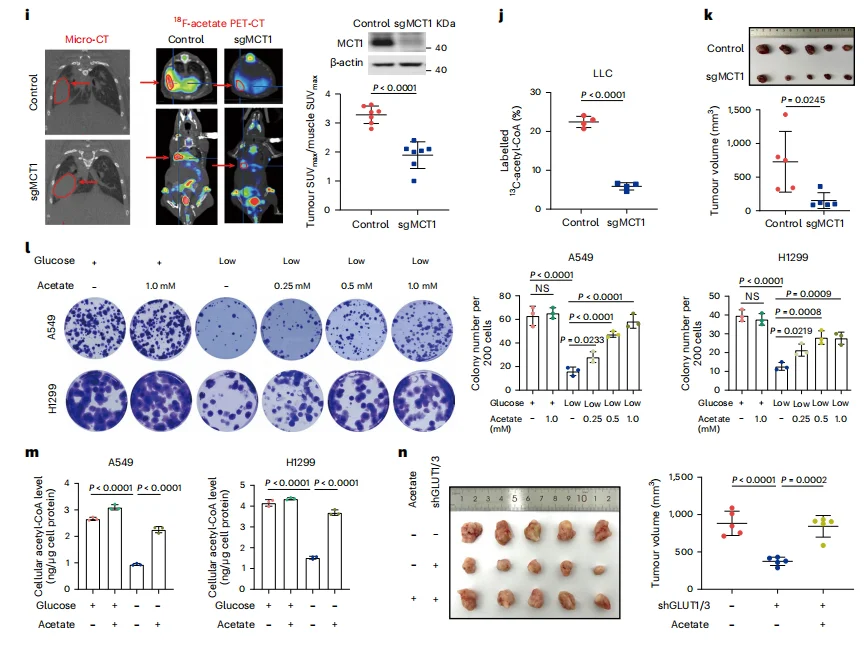
Figure 2. Dietary Acetate Alleviates Energy Stress and Promotes Lung Tumor Growth
-
MCT1 → ACSS2 → DLAT → USP10 → c-Myc Axis Stabilizes the c-Myc Transcription Factor:
Acetyl-coenzyme A (acetyl-CoA) is known to mediate protein acetylation. Under glucose-deprived conditions, supplementation with acetate progressively increased histone acetylation levels over time.Mass spectrometry analysis identified c-Myc as a protein showing significantly enhanced acetylation, whose expression was also regulated by ACSS2, the enzyme catalyzing the conversion of acetate into acetyl-CoA (Figure 3a–c).Further immunoprecipitation of c-Myc revealed that DLAT functions as an acetyltransferase, acetylating c-Myc and concurrently reducing its polyubiquitination (Figure 3e–j). Specifically, acetylation at lysine 148 (K148) of c-Myc critically influenced its stability: the c-Myc p.K148R (acetylation-deficient) mutant and the c-Myc p.K148Q (acetylation-mimetic) mutant exhibited opposite phenotypes in terms of stability and protein levels (Figure 3k–o).These findings indicate that DLAT acts as a bona fide protein acetyltransferase at K148, and under acetate supplementation, it suppresses c-Myc polyubiquitination, thereby enhancing c-Myc stability and expression.
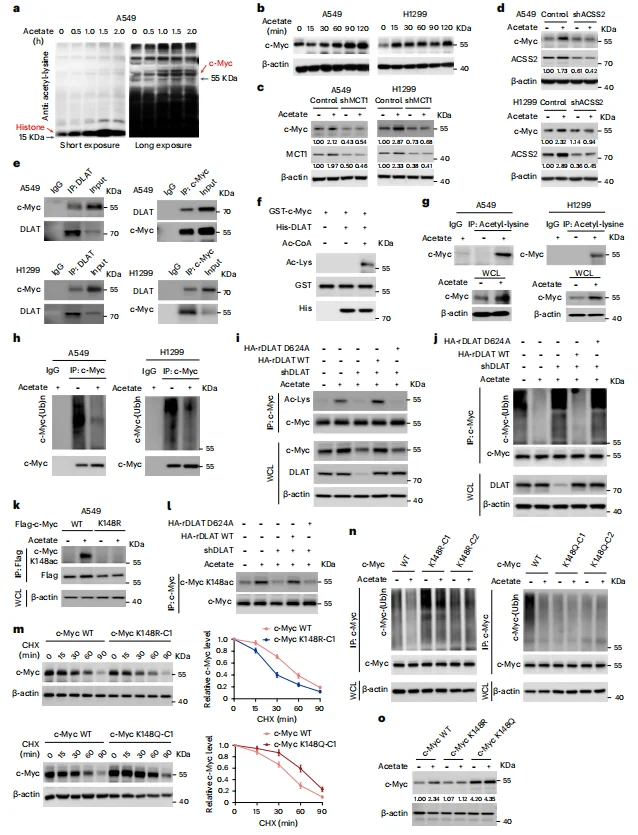
Figure 3. DLAT Acetylates c-Myc at Lys148 and Suppresses c-Myc Polyubiquitination and Degradation
Mass spectrometry analysis and co-immunoprecipitation (co-IP) of anti–c-Myc immunoprecipitates identified USP10 as a c-Myc–binding protein (Figure 4a). Loss of USP10 reduced both basal c-Myc expression and acetate-induced enhancement of c-Myc levels (Figure 4c–e), indicating that USP10 plays a critical role in c-Myc deubiquitination and protection from proteasomal degradation.GST pull-down assays further demonstrated that in the presence of DLAT and acetyl-CoA, wild-type c-Myc (but not the c-Myc p.K148R acetylation-deficient mutant) bound USP10, whereas the c-Myc p.K148Q acetylation-mimetic mutant was able to bind USP10 even without DLAT (Figure 4f).
Together, these results indicate that acetate supplementation promotes DLAT-dependent acetylation of c-Myc at Lys148, which facilitates USP10 recruitment, leading to deubiquitination and stabilization of c-Myc.
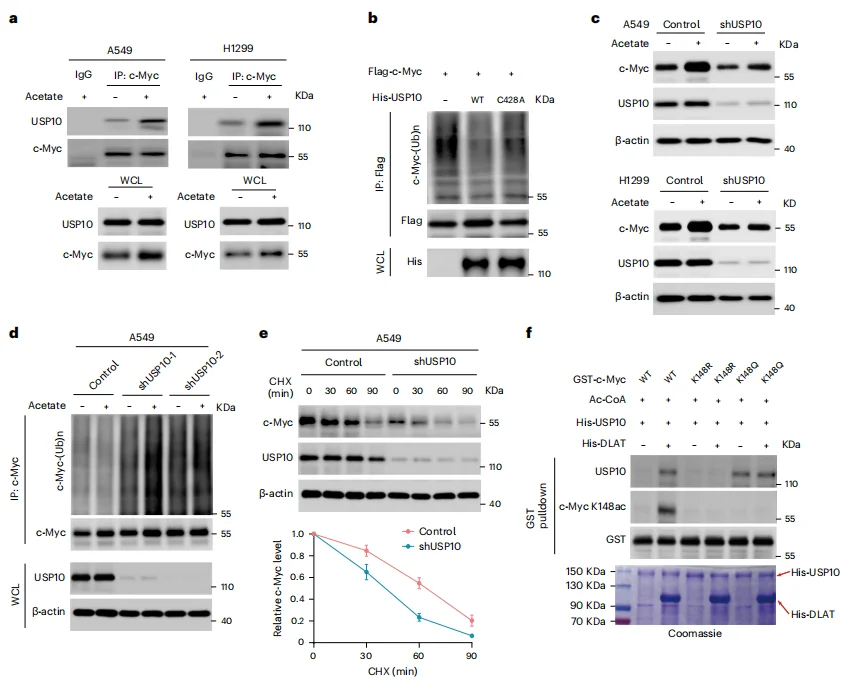
Figure 4. Acetylation of c-Myc at Lys148 Promotes USP10 Binding to Mediate c-Myc Deubiquitination and Stabilization
-
Stabilized c-Myc Promotes Cell Proliferation and Immune Evasion
As a transcription factor, c-Myc orchestrates the expression of multiple target genes. Under glucose-deprived conditions, acetate supplementation significantly increased intracellular levels of c-Myc, PD-L1, cyclin D1, LDHA, and MCT1, while also enhancing lactate production and cellular proliferation (Figure 5b–c).In co-culture with CD8⁺ T cells derived from OT-1 mice, acetate treatment markedly suppressed T-cell activation, reducing IL-2 and IFN-γ production (Figure 5f). Importantly, these effects were abolished by knockdown of MCT1, ACSS2, DLAT, or USP10, or by modulating c-Myc Lys148 acetylation status (Figure 5b–d, g–i).Collectively, these findings demonstrate that acetate supplementation, via the acetate–MCT1–ACSS2–DLAT–USP10–c-Myc axis, promotes acetate uptake, glycolysis, and proliferation of NSCLC cells, while suppressing CD8⁺ T-cell activation in a PD-L1–dependent manner.
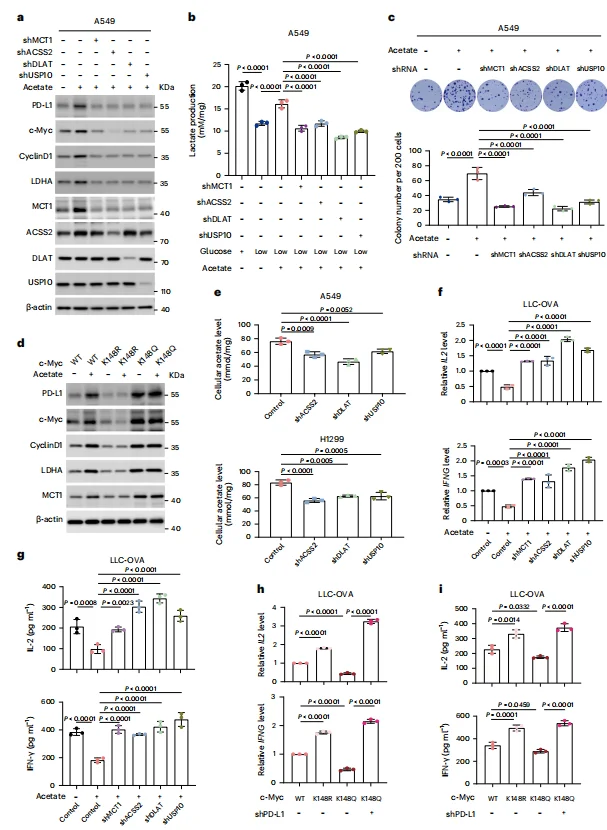
Figure 5. Acetate Supplementation Enhances Glycolysis and Proliferation of NSCLC Cells and Suppresses CD8⁺ T-Cell Activation via a PD-L1–Dependent Pathway
-
c-Myc Lys148 Acetylation–Mediated c-Myc Upregulation Promotes NSCLC Tumor Immune Evasion and Clinical Progression
Immunohistochemical (IHC) analysis of 90 NSCLC tissues and their adjacent normal counterparts revealed that tumor tissues exhibited significantly higher expression of c-Myc, USP10, and c-Myc Lys148 acetylation levels (Figure 6a–d).Consistently, c-Myc Lys148 acetylation levels were positively correlated with MCT1, total c-Myc, and PD-L1 expression (Figure 6e–g), and negatively correlated with CD8⁺ T-cell infiltration (Figure 6h–i).These findings collectively indicate that acetate-driven Lys148 acetylation of c-Myc enhances c-Myc expression, which in turn promotes tumor immune evasion and contributes to NSCLC clinical progression.
Figure 6. c-Myc Lys148 Acetylation–Mediated c-Myc Upregulation Promotes NSCLC Tumor Immune Evasion and Clinical Progression
Significance and Innovations
- Connecting Metabolism and Immunity: For the first time, this study systematically demonstrates how the common metabolite acetate links tumor metabolic reprogramming with immune checkpoint upregulation through a defined molecular pathway.
- Identification of Novel Therapeutic Targets: The work highlights multiple potential targets, including MCT1, ACSS2, DLAT, and USP10. Notably, DLAT’s dual role as a metabolic enzyme and protein acetyltransferase represents a novel discovery.
- Proposing a New Therapeutic Strategy: Limiting dietary acetate intake or pharmacologically blocking acetate utilization may constitute a strategy to “starve tumors” and relieve immune evasion. This approach could be combined with existing immunotherapies (e.g., PD-1 inhibitors) to improve therapeutic outcomes.
Summary
In brief, this study tells the story of a “healthy molecule turned accomplice”: ordinarily benign acetate is hijacked by lung cancer cells, and through a precise MCT1 → ACSS2 → DLAT → USP10 → c-Myc cascade, simultaneously achieves:
· Fueling tumor growth (“charging” cancer cells)
· Promoting immune evasion (“cloaking” cancer cells)
This work not only deepens our understanding of tumor metabolic complexity, but also provides a rational basis and actionable targets for combined metabolic and immunotherapy strategies in lung cancer.
Support Provided by Ubigene
This study utilized A549 cells engineered by Ubigene to carry MYC
(p.K148R) and MYC (p.K148Q) point mutations, enabling mechanistic
dissection of acetate’s role in tumor immune evasion.
For
point mutation cell construction
requirements, Ubigene developed the EZ-HRex™ core technology, which
incorporates the innovative U+ molecule to significantly enhance HDR
efficiency. At the Cell Pool level, HDR genotypes reach up to
84%.
providing robust gene editing outcomes. Ubigene also offers four
flexible point mutation strategies—RNP, pre-screened editors, base
editors, and plasmid resistance methods—covering over 300 cell types to
precisely meet diverse research needs.
Reference
Wang J, Yang Y, Shao F, et al. Acetate reprogrammes tumour metabolism and promotes PD-L1 expression and immune evasion by upregulating c-Myc. Nat Metab. 2024;6(5):914-932. doi:10.1038/s42255-024-01037-4
 Promotions
Promotions 








