CRISPR/Cas9 therapeutics: A cure for Triple-Negative Breast Cancer


CRISPR/Cas9 therapeutics: A cure for Triple-Negative Breast Cancer

Triple-negative breast Xcancer (TNBC) comprises a very heterogeneous group of cancers that lack the receptors like estrogen receptor (ER), progesterone receptor (PR), and HER2/neu that are commonly found in other breast cancers. Among breast cancer patients, TNBC accounts for approximately 15–25% of all breast cancer cases, while the majority of TNBC patients are young women or women with a mutation in the BRCA1 gene. Unfortunately, effective targeted therapies do not exist for TNBC patients, leaving surgery, chemotherapy, and radiotherapy as the only treatment options. The extremely aggressive and metastatic nature of TNBC, coupled with fewer treatment options, has resulted in the worst mortality rates among all breast cancer subtypes, highlighting an urgent and unmet clinical need for novel precision medicines to treat TNBC.
The MDA-MB-231 cell line is a model for human triple-negative breast cancer which exhibits an estrogen-independent state and does not express estrogen receptors. The MDA-MB-231 cell lines are highly invasive and metastatic human breast cancer cells. They display the invasiveness by mediating the proteolytic degradation of the extracellular matrix (ECM), including the basement membrane and several mechanical barriers to the ECM, through the increased expression of matrix metalloproteinases. In vitro cell, migration/invasion assays are a very good indicator of the activity of MDA-MB-231 cells, and western blot for caspase can helps distinguish from other cells. TNBC accounts for 15% to 20% of all breast cancer cases and hard to treat them.
Gene editing technologies are rapidly advancing as a realistic therapeutic option. The ability to strategically edit a patient’s genome can constitute a treatment revolution. Genome editing technologies have huge potential in breast cancer treatment including targeting oncogenes and tumor-suppressor genes, genes related to chemotherapy drug resistance, and genes related to therapies using targeted drugs and inhibitors to promote further preclinical research and the clinical treatment of breast cancer. To date, most studies of CRISPR genome editing therapy have focused on straightforward, monogenic diseases such as cystic fibrosis and hereditary tyrosinemia, and have achieved promising preclinical therapeutic benefits. The therapeutic benefits of in vivo CRISPR genome editing on more complex, multigenic diseases (e.g., TNBC) are still unclear. Using targeted CRISPR genome editing therapeutics to precisely manipulate hereditary or somatic oncogenic mutations in TNBC tumors may bring a paradigm-shifting therapeutic approach for TNBC treatment.
Cell lines and xenograft models are frequently used to study triple-negative breast cancer subtypes in preclinical and translational research. Below, there are some potential applications of MDA-MB-231 cell line
1. Cancer vaccine: Whole-cell vaccine, single tumor antigen-targeted vaccines, autologous tumor cell vaccines, tumor-derived cytokine, immunogenicity, immunosuppressive cytokines, cancer cell-based vaccines, etc. Engineered tumor cells for therapeutic vaccine or preventive vaccine.
2. Critical molecular regulators:Genomics, transcriptomics, proteomics, and immunomes’ for cancer growth, survival, and metastasis.
3. Gene discovery and recurrence: Identify cancer driver genes and uncover cancer-specific vulnerabilities.
4. Drug discovery: Novel therapeutic vulnerabilities and the development of more effective treatments.
5. Disease modeling: Xenograft disease modeling, disease progression, and mechanism studies.
Genome editing is the process of precisely modifying the nucleotide sequence within a gene for wiping out or initiating explicit and preferred characters in the genome. CRISPR/Cas9 is a gene‐editing technology, which can correct errors in the genome and switch on or off certain genes in cells and organisms fast, cheaply, and relatively easily. To date, most studies of CRISPR genome editing therapy have focused on straightforward, monogenic diseases such as cystic fibrosis and hereditary tyrosinemia, and have achieved promising preclinical therapeutic benefits. An MDA-MB-231 cell line is broadly used for human triple-negative breast cancer study (drug-resistant gene, chemotherapy-resistant gene, recurrence, metastasis, drug discovery, disease, and therapeutics). Gene editing within this cell line possible to generate single or multiple gene knockouts, mutation corrections, or insert reporter transgenes. Engineered MDA-MB-231 cell helps investigators to study TNBC breast cancer hallmarks, disclosure of drug resistance mechanism, cancer therapeutics, cell death research, genomics, drug discovery, drug response, and cell therapy. CRISPR/Cas9 technology positively fuel the advancement of in vivo and in vitro gene editing in breast cancer to tackle the complexity of breast cancer metastasis, drug resistance, chemotherapy resistance and have an immense impact on precision medicine. Ubigene developed CRISPR-U™ for gene editing of the MDA-MB-231cell line. Thus, possible to achieve genome-edited cells. Ubigene can customize the gene-editing in eukaryotic cells as well as can generate various genes modification in animal models.
Figure: CRISPR-U™ customized workflow for engineered MDA-MB-231 model cells
CRISPR genome editing in triple-negative breast tumors with nanolipogel
Triple-negative breast cancer (TNBC), which has the highest mortality rate of all breast cancer, is in urgent need of a therapeutic that hinders the spread and growth of cancer cells. CRISPR genome editing holds the promise of a potential cure for many genetic diseases, including TNBC; however, its clinical translation is being challenged by the lack of safe and effective nonviral delivery systems for in vivo therapeutic genome editing. In this study, the researcher reported the synthesis and application of a noncationic, deformable, and tumor-targeted nanolipogel system (tNLG) for CRISPR genome editing in TNBC tumors. The schematic illustration of tNLG structure and in vivo CRISPR genome editing mechanism is shown in Fig1.
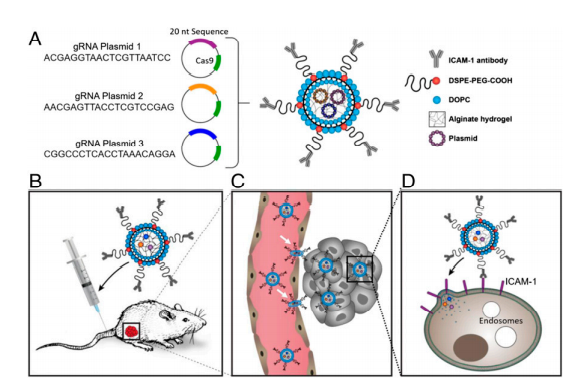
Fig 1: Schematic illustration of tNLG structure and bio mechanisms of in vivo CRISPR genome editing.
The researcher developed engineered tNLG as a CRISPR delivery nanovector verified its applicability in various dimensions like uniformity, core-shell nanostructure, encapsulation efficiencies, storage stability, cytotoxicity (Fig.2a-e). The in vitro TNBC specificity of tNLG in comparison with nNLG, the transendothelial capability of tNLG and tPSNP across the tumor endothelial cell (EC) barrier, and the normal EC barrier (Fig.2f,g). Deformable permeability indicated that tNLG can directly release payload into the cytosol without endosomal entrapment. Fluorescent imaging confirmed that CRISPR plasmids were delivered into the cytosol of TNBC cells by tNLGs. The cell distribution area of internalized tNLGs, indicating that internalized CRISPR plasmids were not confined within endosomes. Plasmids were successfully delivered into the nuclei of MDA-MB-231 cells by engineered tNLG (Fig. 2h-k).
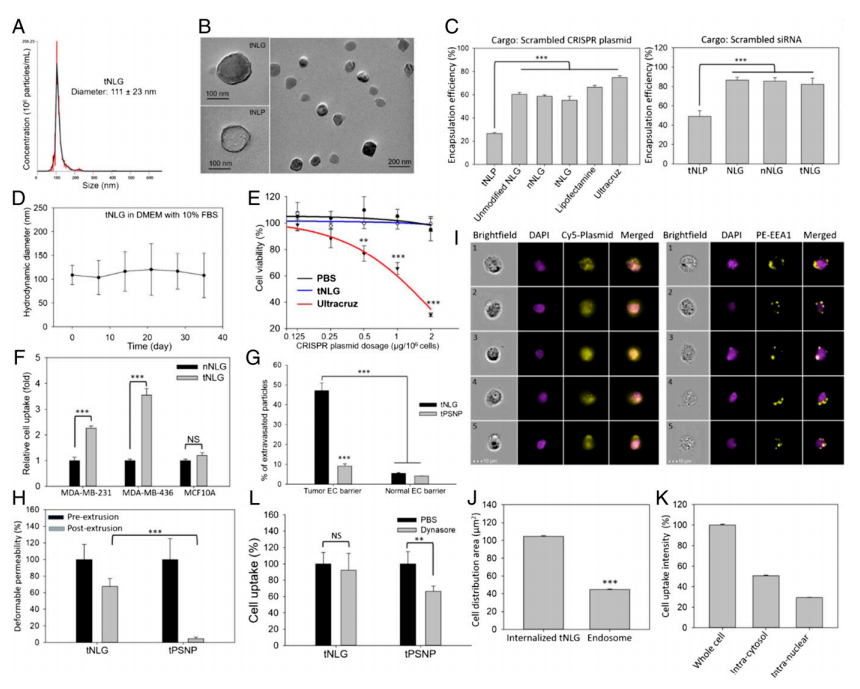
Fig 2: Engineered tNLG as a CRISPR delivery nano-vector.
To demonstrate the therapeutic benefit of CRISPR genome editing, they selected Lcn2 as the therapeutic target for proof-of-principle TNBC-specific genome editing experiments in vitro and in vivo. Lcn2 gene expression was significantly up-regulated in human TNBC cell lines (MDA-MB-231 and MDA-MB-436) in comparison with nonneoplastic MCF10A cells (Fig. 3a,b). TNBC patients with high Lcn2 expression (cohort of 102 patients) demonstrated a significantly worse prognosis than the low Lcn2 group (cohort of 76 patients, P = 0.016; log-rank test). In vitro genome editing efficiency of tNLGs was measured by using qRT-PCR and IF staining. They found that Lcn2 CRISPR knockout in 2 TNBC cell lines did not alter their proliferation. Also, the Lcn2 CRISPR knockout did potently impede cell migration in both MDA-MB-231 and MDA-MB-436 cells (Fig.3c-l).
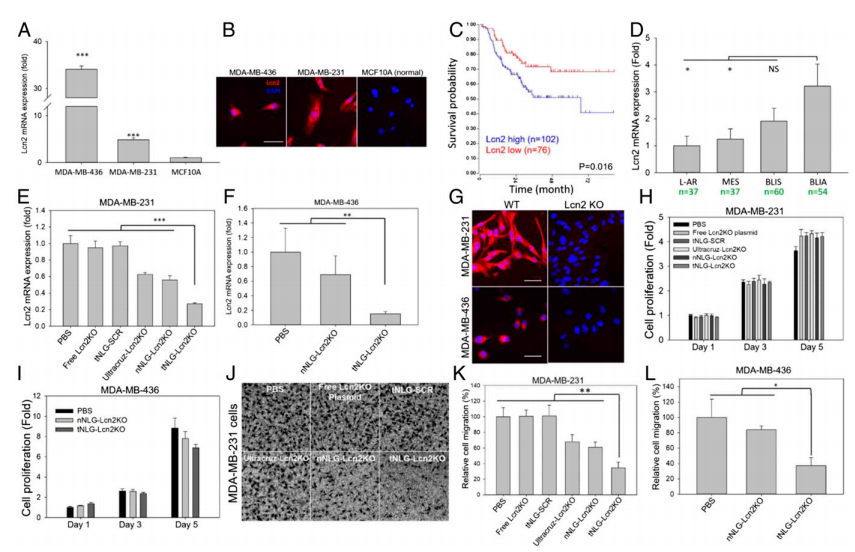
Fig 3: Potent in vitro CRISPR genome editing by tNLG
To evaluate the EMT phenotypical changes caused by Lcn2 CRISPR knockout, the research utilized a state-of-the-art quantitative phase imaging (QPI) method to characterize and compare a panel of cell morphological and behavioral parameters between wild-type (WT) and Lcn2 CRISPR knockout (Lcn2 KO, tNLG-Lcn2KO group) MDA-MB-231 cells. The cell motion trajectories of WT and Lcn2 KO cells are shown in Fig.4a. They found that WT cells had a significantly faster motility speed than Lcn2 KO cells, resulting in much longer migration distances (Fig. 4b-c). Besides, that WT cells exhibited a classic mesenchymal cell phenotype with significantly longer filopodia during cell migration. The Lcn2 KO cells significantly reduced their cell length and cell height resulting in significant inhibition of filopodia formation. Furthermore, Lcn2 KO cells significantly reduced mesenchymal biomarker expression and increased expression of epithelial biomarker (Fig.4d-h). Lcn2 CRISPR knockout in TNBC cells significantly reduces aggressiveness by inhibiting EMT, at least partially, and may lead to a potent in vivo therapeutic benefit in TNBC therapy.
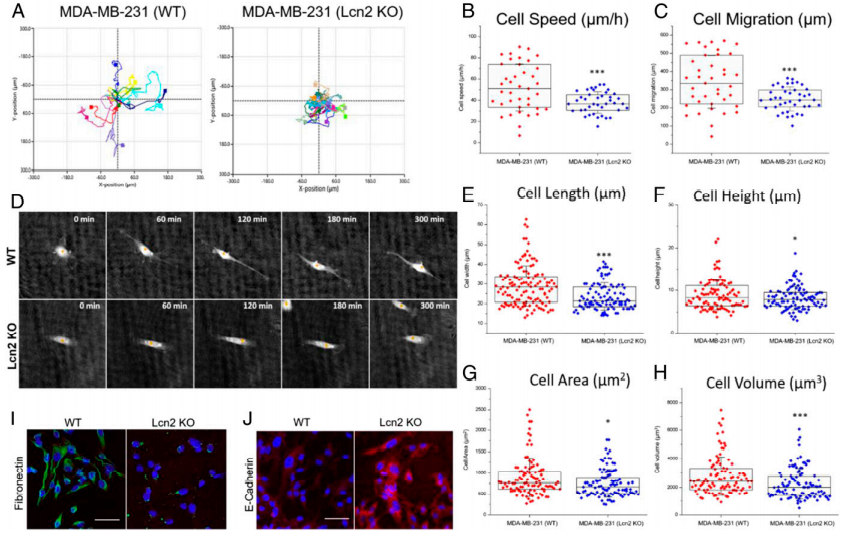
Fig 4: CRISPR genome editing of Lcn2 inhibits EMT of TNBC cells
In vivo therapeutic genome editing in orthotopic TNBC tumors. The experiment was carried out by evaluating the tumor specificity and biodistribution of tNLG and nNLG in an orthotopic TNBC model using in vivo near-infrared (NIR) imaging. The therapeutic efficacy of in vivo CRISPR genome editing using an orthotopic TNBC model (Fig. 5a). Tumor progression was monitored by tumor volume measurement, (Fig. 5b). Tumor mass at endpoint (day 84) was quantified in weight (Fig.5c). Mouse body weights remained unchanged during treatment in all tested groups (Fig. 5e). They quantified the in vivo CRISPR genome editing efficiency by measuring the loss of Lcn2 gene expression in TNBC tumors using qRT-PCR as depicted in Fig. 5f as well as liver and renal toxicities of tNLG-Lcn2KO were determined by measuring serum levels of ALT, AST, Creatinine, and BUN (Fig. 5g).
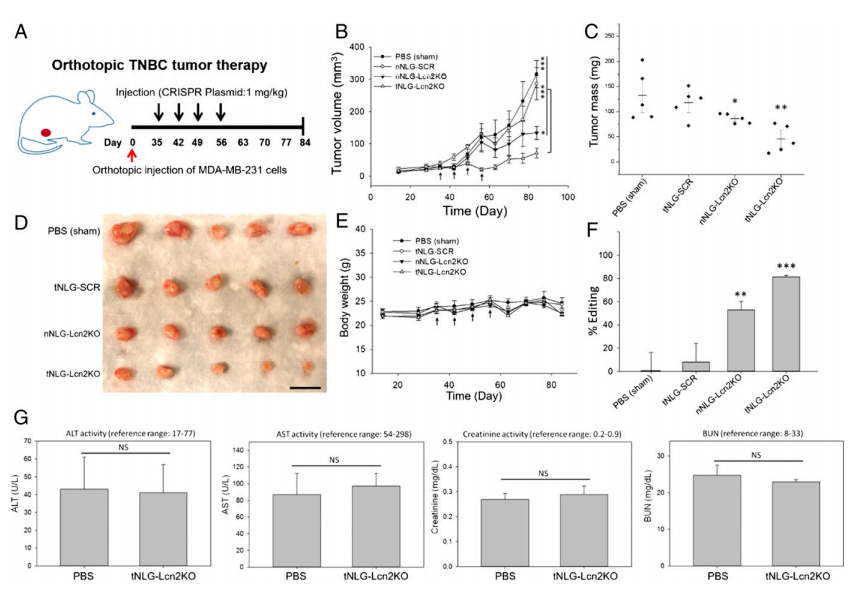
Fig 5: In vivo CRISPR genome editing of Lcn2 potently attenuates TNBC tumor growth
In summary, the researcher developed a noncationic, deformable, and TNBC-specific nanolipogel for in vivo CRISPR genome editing in human TNBC tumors. The tNLGs also represent a platform delivery system that can be used to target TNBC cells. This proof-of-principle study suggests that this tNLG formulation has a promising and broad potential for translating CRISPR genome editing into a novel precision medicine in cancer therapy.
pS134-GR, a potential therapeutic target for aggressive triple-negative breast cancer
Breast cancer (BC) accounts for ~ 15% of cancer-related death in American women. Up to 40% of TNBC tumors express elevated glucocorticoid receptor (GR) levels. GR is a ligand-activated (cortisol/dexamethasone [Dex]) transcription factor member of the steroid hormone receptor (SR) superfamily and its expression associated with chemotherapy resistance and metastatic recurrence of TNBC. Although TNBC is intensely studied, molecular targeted therapies are still largely unavailable for TNBC patients, who suffer from higher disease recurrence, more frequent metastasis, and a worse prognosis. Thus, appropriate biomarkers of driver pathways and new therapeutic targets are urgently needed. Phosphorylation of GR on Serine 134 is elevated in TNBC relative to other breast cancer subtypes. ligand-dependent pS134-GR target genes are known mediators of pro-survival and metastasis in TNBC. In this study, the researcher used CRISPR/Cas9-mediated gene knockin to modify MDA-MB-231 cells to express NR3C1 containing an S134A mutation. This is accomplished using an HDR donor vector along with a CRISPR/Cas9-GFP expression vector; an anti-sense gRNA sequence (NR3C1-S134A-AS1-sgRNA) targeting the coding sequence of NR3C1 was cloned into a CRISPR/Cas9-GFP expression vector (PX458).

Fig : GR Ser134 phosphorylation creates a feedforward signaling loop that potentiates further activation of the p38 MAPK pathway downstream of TGFβ1 in TNBC models.
The investigator tested Dex's role in breast cancer cell migration (Fig.1). MDA-MB-231 cells were treated with increasing doses of Dex at different time points and Dex induces cellular migration was measured (Fig1. b,c). IPA analysis of MDA-MB-231 cells treated with 100 nM Dex and their pathway analysis including the TGFβ1 and p38 MAPK pathways (Fig1. d). The fraction of wound area closure of MDA-MB-231 cells treated (18 h) with vehicle control (Fig1. e).
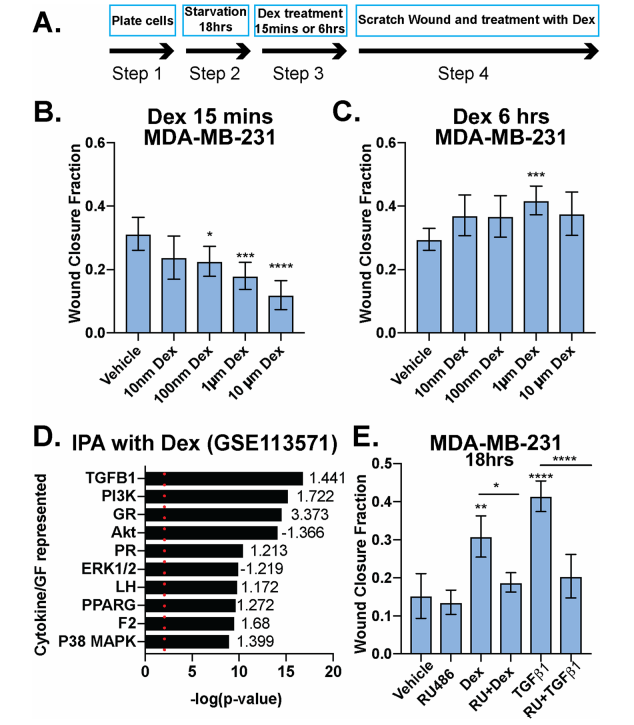
Fig 1: Dexamethasone either inhibits or promotes breast cancer cell migration in a time-dependent manner
The researcher studied the linkage between TGFβ1 signaling and phosphorylation of GR Ser134 in MDA-MB-231 cells and they found that TGFβ1 induces p38 MAPK-dependent phosphorylation of GR Ser134 shown in Fig 2a,b, and c. A Patient-derive xenograft (PDX) HCI-10 cell line was treated with TGFβ1 and evaluated the expression of pS134-GR, total GR, and total p38 (Fig.2d). Western blot analysis of pS134-GR, total GR, p-p38 MAPK, and total p38 MAPK protein levels in MDA-MB-231 cells treated with either vehicle control or HGF shown in Fig. 2e.
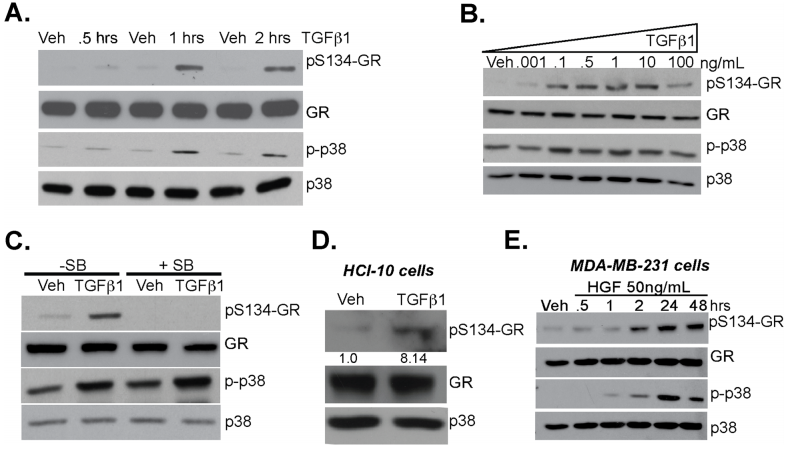
Fig 2: TGFβ1 induces p38 MAPK-dependent phosphorylation of GR Ser134
To test the requirement for GR Ser134 in TGFβ1- regulated TNBC cell migration, they employed a CRISPR/ Cas9 approach to creates MDA-MB-231 cells expressing either wt-GR (control) or a point mutant GR in which Ser134 has been changed to Alanine (S134A-GR clone #1 and clone #2). They assessed MDA-MB-231 cells and their clone for phosphorylation, MTT, wound healing, migratory activity, and tumorsphere assay (Fig.3 a-f). Impaired cell migration measured by chemotaxis in transwell migration assays (Fig. 3g), as well as attenuated tumorsphere formation, were also observed using the previously described GRnull/low U2OS osteosarcoma cell line engineered to stably express S134A-GR relative to wt-GR (Fig. 3h).
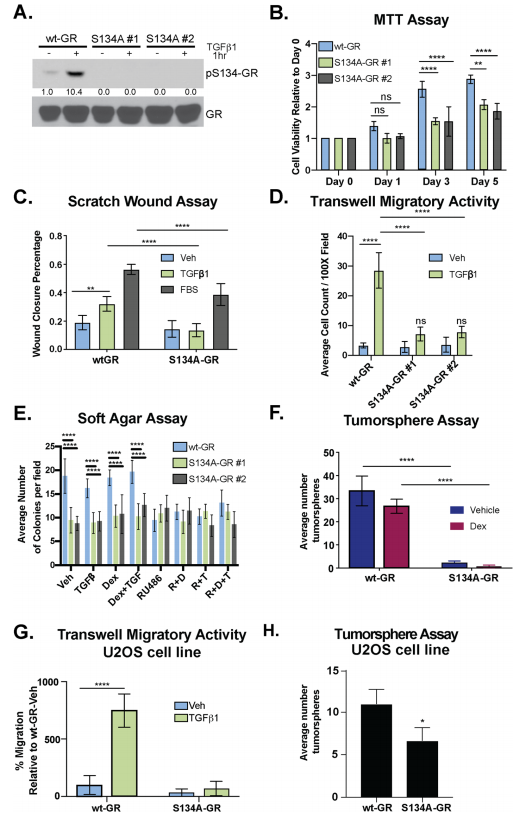
Fig 3: GR Ser134 is required for TGFβ1-mediated migration of TNBC cells
The expression of 14-3-3ζ mRNA in patients with ER−/HER2- (TNBC) or Her2+ breast cancer was higher when compared to patients with ER+/HER2- breast cancer (Fig. 4a) and higher expression correlates with poor survival (Fig.4b). Besides, they observed elevated expression of 14-3-3 in different breast cancer cell lines including MDAMB-231, Hs578T, and MDA-MB-468 (TNBC) cells (Fig.4 c). They tested the necessity for 14-3-3ζ in TGFβ1-mediated migration and found that TGFβ1 induced robust cell migration in sh control but not sh14-3-3ζ TNBC models. Western blot analysis was employed to evaluate the interaction of 14-3-3ζ with GR (Fig.4d,e).
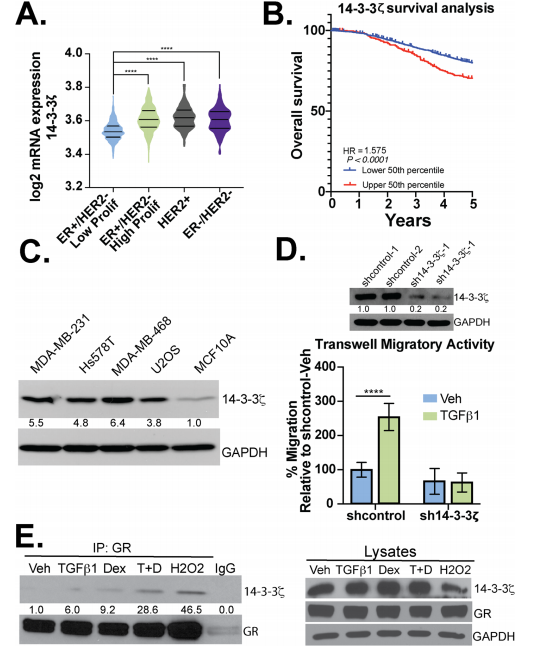
Fig 4: 14-3-3ζ is required for TGFβ1-induced TNBC cell migration
The impact of pS134-GR on TNBC cell behavior was performed by RNA-seq studies in MDA-MB231 cells expressing either wt-GR or S134A-GR (clone #1). RNA-seq data shown the differences in transcriptomes between MDA-MB-231 cells expressing either WT or S134A-GR cells shown in Fig 5.
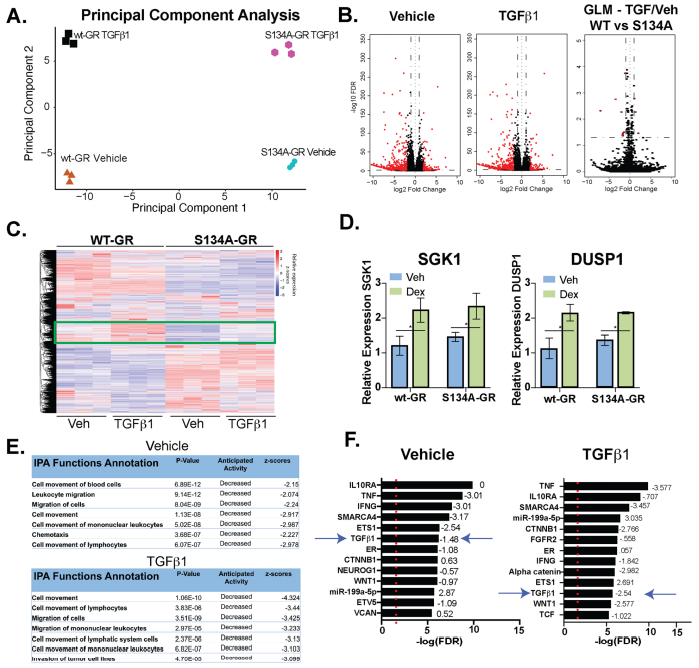
Fig 5: WT vs. S134A-GR transcriptomes in TGFβ1-treated MDA-MB-231 cells
To study phosphorylation of GR Ser134 is critical for MAPK signaling, they reveal a dramatic loss of expression of genes important for MAPK signaling in TNBC cells expressing S134A-GR compared to wt-GR (Fig. 6a). Western blot analysis of different protein expression shown in knock-in, point mutation, and knockdown cell line (Fig. 6b,c,d). They also found that MAPK3K5 protein levels were significantly downregulated in two clones harboring S134A-GR relative to wt-GR (Fig.6e). MAP3K5 and GR mRNA expression levels are strongly correlated in TNBC patients (Fig.6f). Notably, inhibition of MAP3K5 with the selective inhibitor, selonsertib, blocked phosphorylation of GR (Fig. 6g) and inhibited TGFβ1-induced migration (Fig. 6h) in both MDA-MB-231 and Hs578T cells. Similarly, inhibition of p38 MAPK using SB203580, halted TGFβ1-mediated migration in both TNBC models (Fig. 6h).
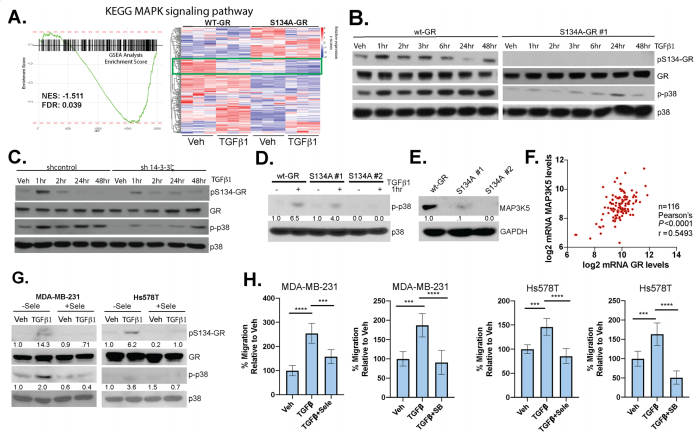
Fig 6: Phosphorylation of GR Ser134 is critical for MAPK signaling
The researcher found that P-S134-GR promotes the expression of a 24-gene signature, these are correlated with poor prognosis in Breast cancer. Proteins upregulated in MDA-MB-231 cells expressing either wt-GR or S134A-GR shown in Fig7a. Gene Clustering of the 39 genes significantly upregulated in wtGR cells by TGFβ1 (Fig.7b). Representative genes (LEFTY2, PIK3IP1) were validated by qPCR in TGFβ1-treated cells predicted by RNA-seq data. They found that recruitment of S134A GR to these regions was significantly diminished (LEFTY2) or failed to occur (PIK3IP1) in CRISPR models expressing phosphomutant GR (Fig. 7d). To further evaluate the importance of the pS134-GR gene signature in breast cancer patients, they used the METABRIC dataset to calculate the average expression of the above-defined 24 pS134-GR-induced genes for each patient tumor and stratified patient populations. This difference was significant with a log-rank p-value of 0.0008. These findings in the METABRIC dataset were verified by using the SurvExpress tool with the TCGA breast cancer dataset and observed similar results with significant separation based on overall survival (Fig. 7e, f).
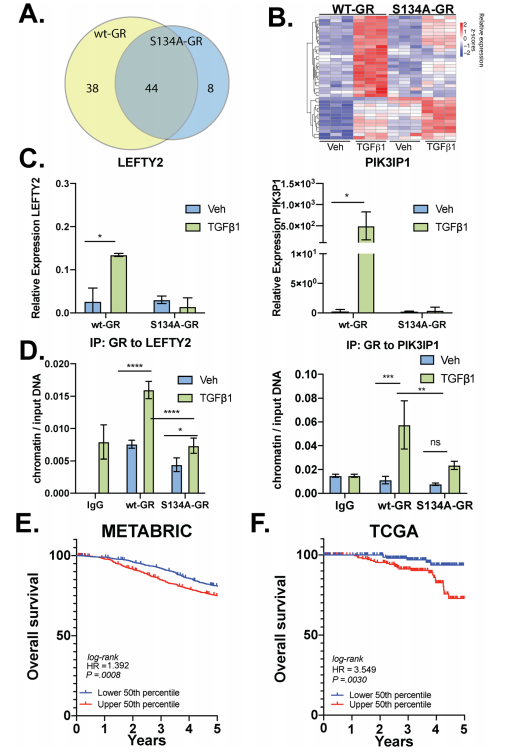
Fig 7: P-S134-GR promotes the expression of a 24-gene signature that correlated with poor prognosis in BC
In summary, phospho-GR is a key mediator of dangerous TNBC progression. Ligand-independent but p38 MAPK-induced phosphorylation of GR on Ser134 is essential for its deleterious actions as a driver of TNBC migration, invasion, anchorage-independent cell growth, and tumorsphere formation.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!

Reference:
Therapeutic genome editing of triple-negative breast tumors using a noncationic and deformable nanolipogel. PNAS, 2019, 116 ( 37)18295–18303.
Glucocorticoid receptors are required effectors of TGFβ1-induced p38 MAPK signaling to advanced cancer phenotypes in triple-negative breast cancer. Breast Cancer Res. 2020, 22(1):39.








