Expert Insights | Tips for EO771 Cell Culture and Gene Editing


Expert Insights | Tips for EO771 Cell Culture and Gene Editing

As a classic breast cancer model derived from C57BL/6 mice, EO771 cells have become a star cell line in popular fields such as breast cancer immunotherapy, immune checkpoint inhibition research, and gene editing modeling, thanks to their excellent immunocompatibility and high sensitivity to the tumor immune microenvironment. From functional validation of CAR-T therapy to the establishment of CRISPR high-throughput screening platforms, EO771 consistently proves to be the most reliable tool in the hands of researchers, aiding you in exploring the cutting-edge intersection of tumors and immunity. Today, Ubigene will comprehensively unlock the usage secrets of EO771—sharing cell culture experiences summarized by frontline researchers and efficient gene editing techniques to help you achieve more with less effort and easily excel in your experiments.
Order EO771 cell now>>>Cell Information
Cell Name: EO771 (Murine Medullary Breast Cancer Cell Line)
Cell Morphology: Epithelial-like, adherent
Cell Culture Conditions: 90% DMEM + 10% FBS
Gas Phase: Air 95%; CO2,5%
Temperature: 37°C
Media Change Frequency: Every 2-3 days
Passage Ratio: 1:2 - 1:4
Normal Cell Growth Image: Grown in an adherent manner, forming a single layer, typically polygonal or spindle-shaped, with clear cell boundaries
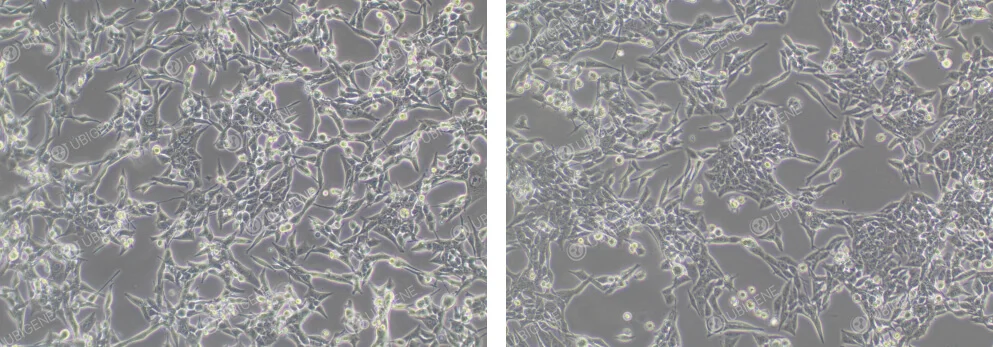
Figure 1. Normal EO771 Cell Growth
Poor Cell Growth Image: Cell morphology changes, with cells becoming rounder and more floating. The cytoplasm may vacuolate, and cells may collapse, losing their three-dimensional structure; signs of aging are present
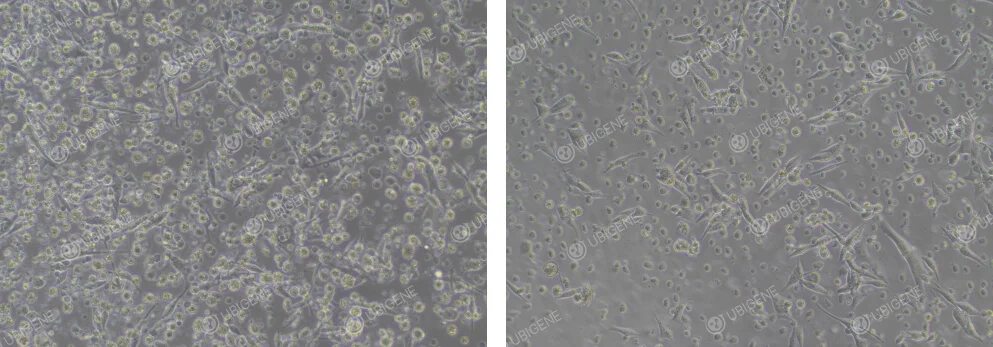
Figure 2. Poor EO771 Cell Growth
Cell Thawing
1)Preparation: Prepare 7 mL of complete culture medium in a centrifuge tube.
2)Thawing: Quickly take the cells from dry ice, hold the lid with tweezers, and place them into a 37℃ water bath. Rapidly shake until fully thawed (note: water should not cover the lid). In about 1 minute, it would completely thaw.
3)Centrifugation: Transfer the thawed cell suspension to a centrifuge tube and centrifuge at 1100 rpm for 4 minutes. Discard the supernatant.
4)Resuspension: Resuspend the cells in complete culture medium and transfer the cells into appropriately sized culture dishes or flasks.
5)Culturing: Place the culture dishes or flasks in a 37℃ incubator and observe the cell condition and adhesion after 24 hours.
Cell Passaging (Using T25 flasks as an example)
1)When cells reach 80-90% confluency, passage them by discarding the culture medium in a clean bench and washing the cells with 5 mL PBS 1-2 times.
2)Add 1 mL trypsin, gently shake the culture flask to ensure trypsin completely soaks the cells, then place the culture flask in the incubator for 1-3 minutes. When observing under a microscope, if most cells appeared rounded and bright, and the cells detached when gently shake the culture flask, immediately stop digestion.
3)Add 2 mL of complete culture medium (double the trypsin volume) to stop digestion, then transfer the mixture to a 15 mL centrifuge tube.
4)Centrifuge at 1100 rpm for 4 minutes at room temperature. Discard the supernatant, and resuspend the cells in complete culture medium.
5)Passage the cells in a ratio of 1:2 to 1:4, and observe the cell condition the next day.
Cell Cryopreservation
1)Cell Collection: Follow the cell passaging process to collect the digested cells into a centrifuge tube.
2)Centrifugation: Centrifuge at 1100 rpm for 4 minutes and discard the supernatant.
3)Resuspension and Cryopreservation: Resuspend the cells in cryopreservation solution, distributing them into cryogenic tubes at a density of 1×10^6 cells/mL, and label with name, passage number, and date.
4)Storage: Place the cryogenic tubes in a controlled-rate freezing container, store them overnight at -80℃, then transfer them to a liquid nitrogen tank for preservation.
Please Note: These cells grow rapidly, it is necessary to adjust the passage ratio accordingly. Pre-warm the culture medium before changing the medium to prevent cell shrinkage. If cell shrinkage occurs, consider a 1:1 passage ratio after digestion.
General Suggestions for Adjusting Cell Conditions
1)Culture Medium and Serum: Ensure the correct basal medium is used and adjust the serum concentration appropriately based on cell condition.
2)Culture Environment: Check if the cultivation temperature, humidity, and gas phase conditions are normal.
3)Avoid using expired or long-stored media: Freshly prepared complete media should be used within two weeks.
4)Cell Passaging Procedures: During passage, monitor digestion time and trypsin concentration to avoid cell damage caused by prolonged or insufficient digestion.
5)Investigate whether over-digestion or abnormal pH values of culture medium could be affecting the cells, adjust digestion time and pH accordingly.
6)Cell clumping: Gently pipette after digestion (using a pipette tip to repeatedly aspirate and dispense until a single-cell suspension is obtained). Try Accutase to replace trypsin for gentler dissociation.
Suggestions for Adjusting Cell Senescence
1)Confirm that the cultivation conditions and environment are correct.
2)Avoid Over-Digestion: Use an appropriate trypsin concentration and digestion time; Avoid forceful pipetting to prevent cell damage.
3)Increase Serum Concentration: Choose high-quality fetal bovine serum to support cell health.
4)Adjust Cell Density: High cell density may lead to nutrient deprivation and waste accumulation, accelerating cellular aging; Maintain cell density within suitable ranges.
5)Add Growth Factors: Adding growth factors can promote cell proliferation and help ameliorate aging states.
6)Change to Low-Passage Cells: Use low-passage cells to ensure optimal growth and function.
7)Isolate clonal cells with growth advantages from aging cell populations using clonal methods and cultivate them for purifica.
Suggestions for for EO771 Cell Transfection
1)Ensure cells are in good condition and in the logarithmic growth phase, with optimal cell density typically between 70-80%.
2)Monitor digestion time to avoid excessive cell damage.
3)Ensure cells are dissociated into single cells to avoid clumping.
4)Ensure cell viability is ≥80% during experiments.
5)For electroporation, control the number of cells and seed them into appropriate culture plates.
6)Post-electroporation, ensure an adhesion rate of ≥70%.
7)Control experimental timings during electroporation; The procedure should not be excessively prolonged.
8)Cell state may worsen after electroporation; Increase the serum concentration in the culture medium to improve cell recovery.
9)For lentivirus applications, maintain cell confluency at 30-40% prior to infection, to prevent excessive density.
10)Prior to formal experiments using viral methods, conduct preliminary tests to determine the most suitable MOI; add agents such as Polybrene before transfection.
11)Ensure transfection reagents are fully mixed before use to ensure homogeneity.
12)It is recommended to conduct preliminary drug screening tests to find the optimal drug concentrations before proceeding with full-scale screenings post-transfection.
Suggestions for EO771 Single-cell Cloning Experiment
1)Ensure cells are in good condition, with few senescent cells, and maintain approximately 70% confluency before expansion.
2)Ensure cell viability is ≥85% for seeding clones.
3)Conduct preliminary experiments to determine an appropriate clonal seeding density, avoiding too low a proportion of single clones.
4)Ensure even distribution of cells when seeding into 96-well plates.
5)When diluting cell counts, aim for concentrations between 1×10^6 and 2×10^6.
Image of Cells After Electroporation
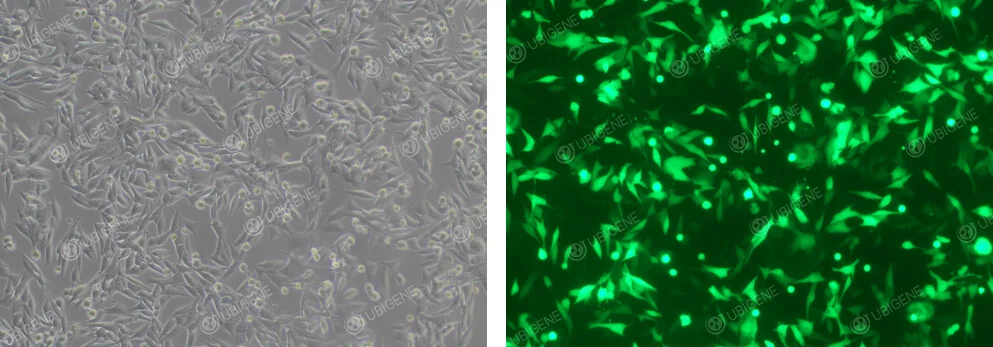
Figure 3. EO771 Cells After Electroporation
Image of Cells After Lentiviral Infection
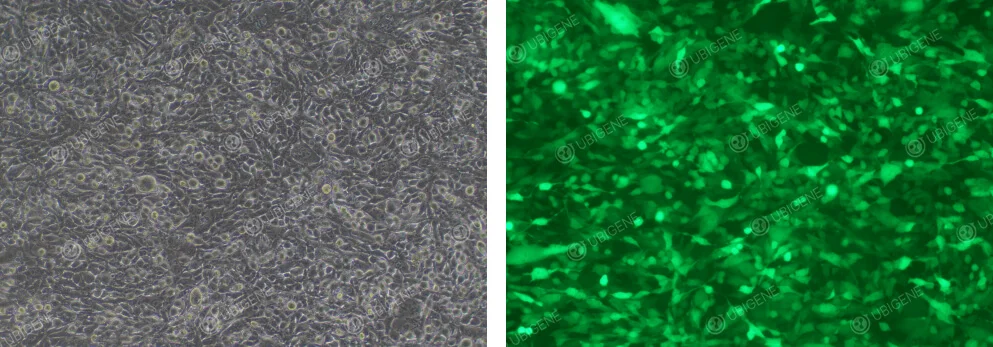
Figure 4. EO771 Cells After Lentiviral Infection








