Single-Dose Genome Editing Therapy Restores Auditory and Vestibular Function in Adult Mice with DFNA41 Hearing Loss


Congenital hearing loss is one of the most common birth defects worldwide. In developed countries, the incidence of congenital sensorineural hearing loss due to genetic factors reaches 1 in 500 newborns. Although over 150 deafness-associated genes have been identified, effective biologic therapies for hereditary hearing loss remain extremely limited. Current clinical interventions, such as hearing aids and cochlear implants, have significant limitations—they offer poor speech recognition in noisy environments, suboptimal music perception, and cannot fundamentally restore the functional deficits of damaged inner ear cells. This highlights an urgent need for novel curative strategies.
In August 2025, Dr. Zheng-Yi Chen's team at Mass Eye and Ear, Massachusetts, USA, published their latest study in the Journal of Clinical Investigation. Using P2rx2^V61L/+ mice (a model of human DFNA41), the team developed an AAV2-mediated SaCas9-sgRNA genome editing system, targeting the adult inner ear for efficient and safe gene correction. Their study demonstrated rescue of auditory and vestibular function, as well as reduction of noise sensitivity in adult mice. Furthermore, the team screened genome editing tools for the human P2RX2 V60L mutation, laying the groundwork for future clinical translation.

Research Background
Gene therapy, including genome editing approaches, offers new hope for treating hereditary hearing loss. Over the past decades, multiple strategies—such as gene replacement, gene augmentation, gene silencing, and genome editing—have been developed and demonstrated partial success in mouse models. However, existing studies face two major limitations:
1.Timing of intervention: Most studies focus on neonatal mice, whose inner ears are still immature. In contrast, the human neonatal inner ear is fully developed, and the efficacy of interventions in the adult inner ear remains largely untested across most gene models. To date, only three mouse models— Otof, Tmprss3, and Mir96 —have demonstrated successful adult-stage interventions.
2.Limited disease coverage: Research on autosomal dominant hearing loss, such as DFNA41, is relatively scarce. These conditions often have delayed onset and progressive deterioration, frequently accompanied by vestibular dysfunction and heightened sensitivity to noise, for which current therapies are inadequate.
DFNA41, a prototypical autosomal dominant progressive hearing loss, is caused by mutations in the P2RX2 gene (e.g., the human V60L variant). Patients typically develop symptoms during adolescence, with hearing progressively declining with age, and exhibit high susceptibility to noise-induced hearing loss (NIHL). Additionally, P2RX2 dysfunction can affect vestibular function, though in humans these deficits are often subtle or compensated by central mechanisms, and thus frequently go unrecognized.
Research Highlights
1.Development of a Highly Efficient, Allele-Specific Editing System with Minimal Off-Target Effects
To achieve precise targeting of the P2rx2^V61L mutant allele, the research team compared the efficiency and specificity of two CRISPR-Cas systems: Staphylococcus aureus Cas9 (SaCas9) and Streptococcus pyogenes Cas9 (SpCas9).
- Editing efficiency and specificity: In primary fibroblasts derived from P2rx2^V61L/+ mice, SaCas9-sgRNA1 achieved an editing efficiency of 75.01% ± 4.55% on the mutant allele, while sparing the wild-type allele (0.45% ± 0.39%). In contrast, SpCas9-based sgRNAs, although editing the mutant allele at 71.28%-83.29%, also showed substantial editing of the wild-type allele (8.73%-29.20%), posing significant off-target risk.
- Indel profile and therapeutic mechanism: The indels generated by SaCas9-sgRNA1 were predominantly single-nucleotide deletions, with 85.1% causing frameshift mutations. This leads to premature termination of P2rx2^V61L translation, producing truncated, non-functional protein and effectively abolishing the gain-of-function effect of the mutant allele, consistent with the therapeutic rationale for DFNA41.
- Genome-wide off-target assessment: Using CIRCLE-Seq, no unintended indels were detected outside the target site. Further NGS validation of seven high-risk, bioinformatically predicted off-target loci showed indel rates <0.1%, confirming that the SaCas9-sgRNA1 system exhibits exceptional genome-wide specificity, addressing a core safety concern in therapeutic genome editing.
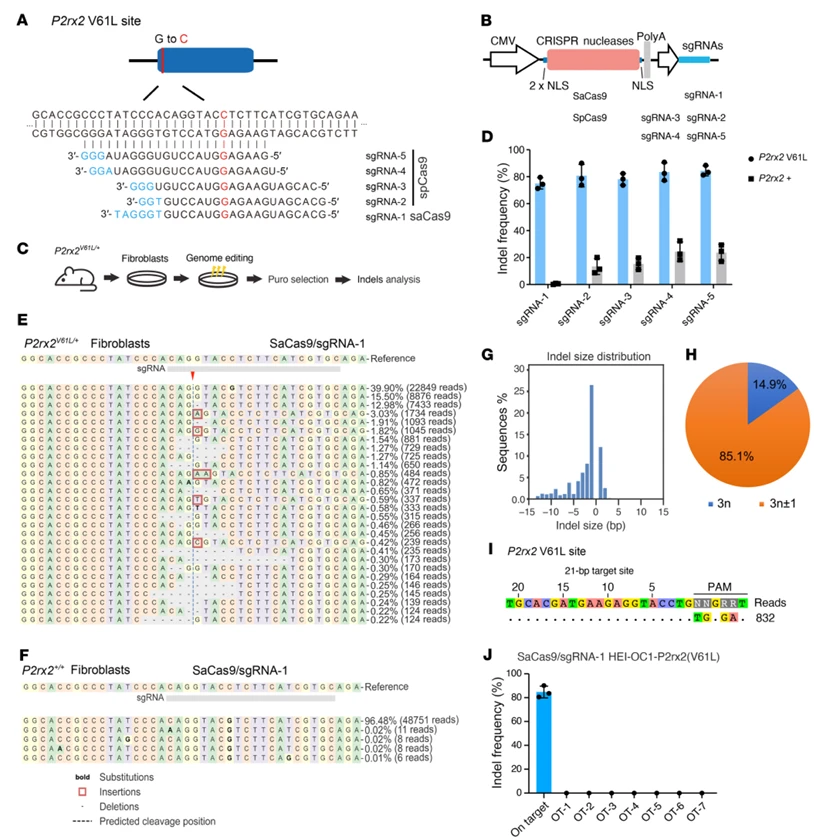
Figure 1. Allele-Specific Genome Editing in Primary P2rx2^V61L/+ Mouse Cells Using SaCas9-sgRNA1
2.Efficient and Safe In Vivo Editing in the Mature Cochlea of Adult Mice
To address the challenge of limited intervention efficacy in the mature human inner ear, the research team delivered AAV2-SaCas9-sgRNA1 to the cochlea of 4-week-old P2rx2^V61L/+ mice (inner ear fully matured) via a round window membrane + cochlear fenestration (RWM+CF) injection. Molecular analyses confirmed both editing efficacy and safety:
- Genomic editing: Eight weeks post-injection, the indel rate at the mutant P2rx2^V61L allele in cochlear tissue was 2.62% ± 0.64%, with no detectable editing of the wild-type allele.
- Transcript-level effect: The proportion of unedited mutant transcripts relative to wild-type transcripts decreased by 28.2%, with only 1.46% exon 2-skipped transcripts detected, which are predicted to have negligible functional impact.
- Hair cell–specific editing: Using FM1-43FX labeling to isolate cochlear hair cells (the primary site of P2rx2 expression), the mutant allele indel rate reached 26.96% ± 4.2%, consistent with transcript-level measurements. No AAV genome integration was detected, and SaCas9 mRNA was undetectable 12 weeks post-injection, minimizing the risk of immune responses.
- Transduction efficiency: AAV2 achieved 100% transduction of inner hair cells across all cochlear turns, while outer hair cells showed a gradient from apex (80%) to base (65%). Transgene expression in inner hair cells was uniform.
- Safety evaluation: Administration of 4*10⁹ vg per ear did not alter auditory brainstem response (ABR) or distortion product otoacoustic emission (DPOAE) thresholds in wild-type mice, confirming that the delivery method does not impair normal hearing.
These results demonstrate that AAV2-SaCas9-sgRNA1 enables efficient and safe in vivo genome editing in the mature cochlea of adult mice, validating its potential for therapeutic intervention in postnatal hearing loss.
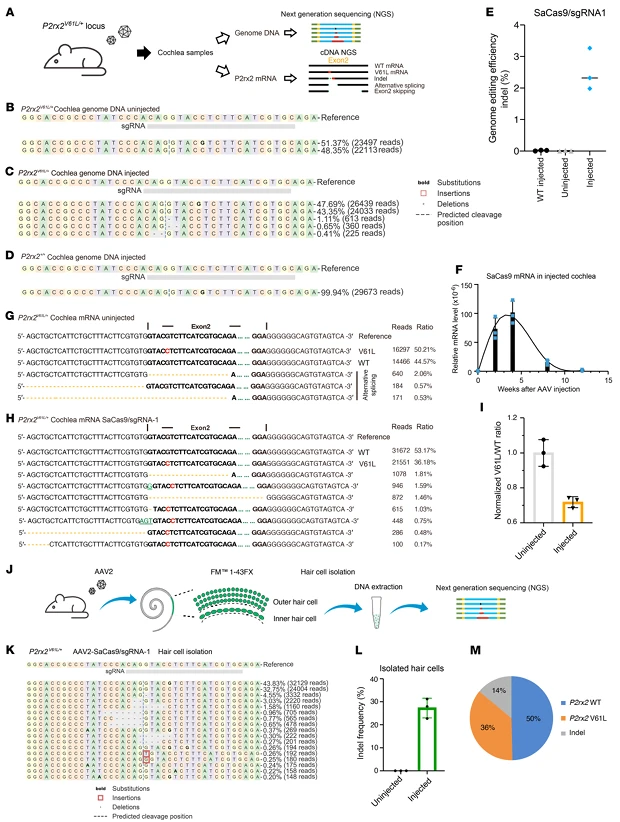
Figure 2. AAV2-Mediated Genome Editing of the P2rx2^V61L Locus in the Cochlea of Adult P2rx2^V61L/+ Mice
3.Function and the Advantage of Early Intervention
The research team systematically evaluated the impact of genome editing on auditory function using auditory brainstem response (ABR) and distortion product otoacoustic emission (DPOAE) measurements, revealing that the timing of intervention significantly influences the extent of protection:
- Adult intervention (4-week-old mice): Within 9 months post-injection, ABR thresholds at 5.66, 8.00, and 11.32 kHz decreased by an average of 13–21 dB, while DPOAE thresholds at 11.32 kHz (reflecting outer hair cell function) decreased by 7–11 dB. Additionally, ABR P1 amplitudes increased and latencies shortened, approaching wild-type auditory conduction characteristics, demonstrating that editing provides long-term, stable preservation of auditory function.
- Early intervention (2-week-old mice, pre-onset of hearing loss): ABR threshold rescue extended up to 16 kHz, with protective effects lasting 12 months. Even at 12 months, ABR thresholds at 8.00 and 11.32 kHz remained 16 dB lower on average. Mechanistic studies suggest that the advantage of early intervention stems from the presence of intact high-frequency hair cells in juvenile mice, which are more amenable to functional restoration via genome editing. These results provide experimental support for the clinical principle of "early diagnosis, early intervention."
- Safety in wild-type mice: Injection of the editing system into 2-week-old wild-type CBA mice did not affect ABR or DPOAE thresholds over 12 months, confirming that the genome editing tools do not interfere with normal hearing.
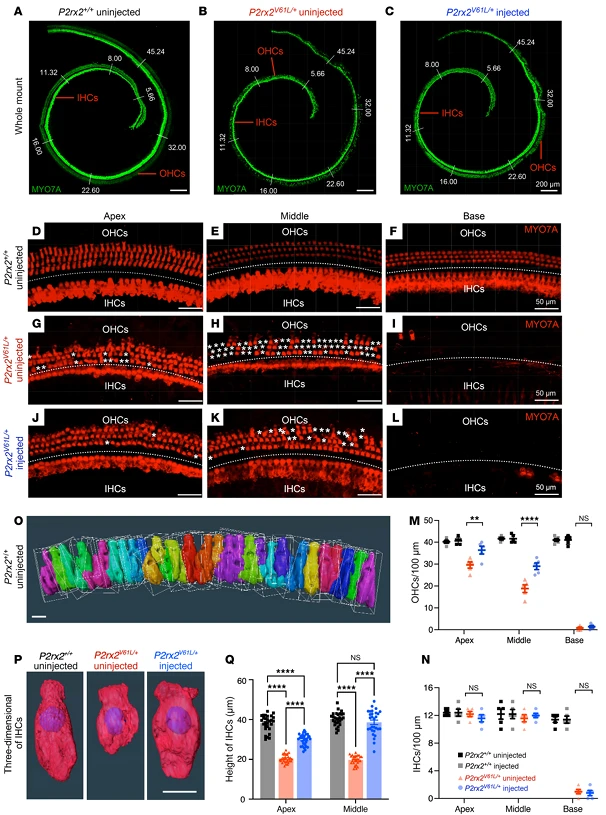
Figure 3. V2–SaCas9–sgRNA1 Adult Injection Restores Outer Hair Cell (OHC) and Inner Hair Cell (IHC) Length in the DFNA41 Mouse Model (P2rx2^V61L/+)
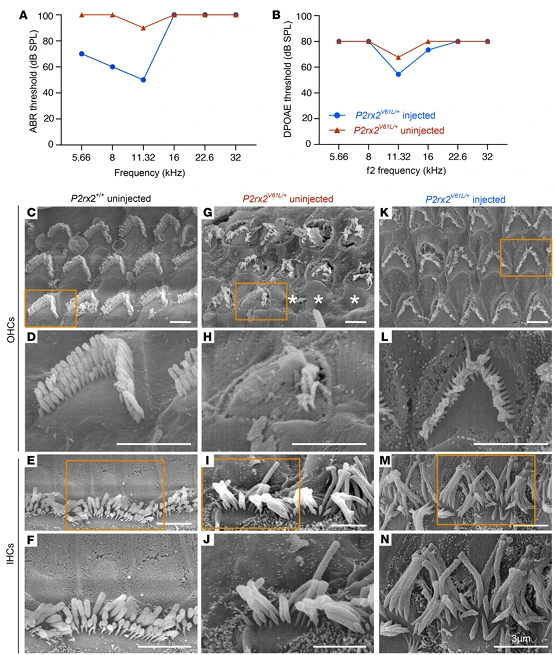
Figure 4. AAV2–SaCas9–sgRNA1 Rescues Hair Cell Morphology in the DFNA41 Mouse Model (P2rx2^V61L/+)
4.Sensitivity and Restoration of Vestibular Function
Beyond auditory function, the study evaluated the impact of genome editing on other key DFNA41 phenotypes:
- Noise sensitivity: Juvenile mice (2-week-old) injected with the editing system were exposed to 97 dB SPL noise (1–20 kHz, 2 hours). ABR thresholds showed no difference compared with non-exposed controls, and inner hair cell ribbon synapse (CtBP2-labeled) loss was fully rescued. In adult mice (4-week-old) injections partially mitigated noise-induced damage, significantly reducing ABR threshold shifts at 8.00 and 11.32 kHz without affecting outer hair cell function, addressing the clinical issue of hyperacusis in DFNA41 patients.
- Vestibular function: Nine months post-editing, the number of surviving utricle hair cells increased by ~30% compared with untreated mice, approaching wild-type levels. Behavioral assays demonstrated restoration of normal activity: hyperactivity and whole-body spinning in the open-field test disappeared, with movement distance and speed returning to normal. In the rotarod test, balance retention was significantly improved.
This study provides the first evidence that genome editing can correct vestibular deficits associated with hereditary deafness, offering new avenues for clinical evaluation and treatment of vestibular dysfunction in DFNA41 patients.
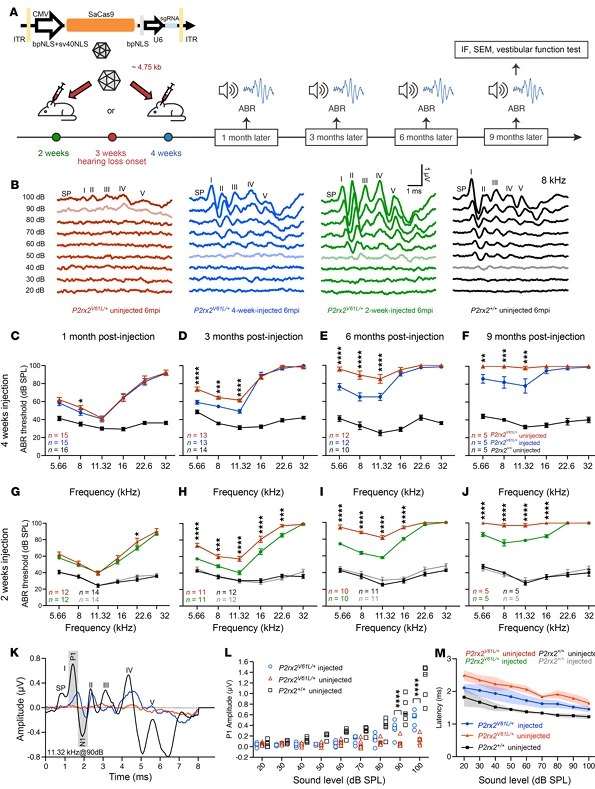
Figure 5. AAV2-SaCas9-sgRNA1 Injection in Juvenile and Adult Mice Sustains Auditory Function in the DFNA41 Mouse Model (P2rx2^V61L/+)
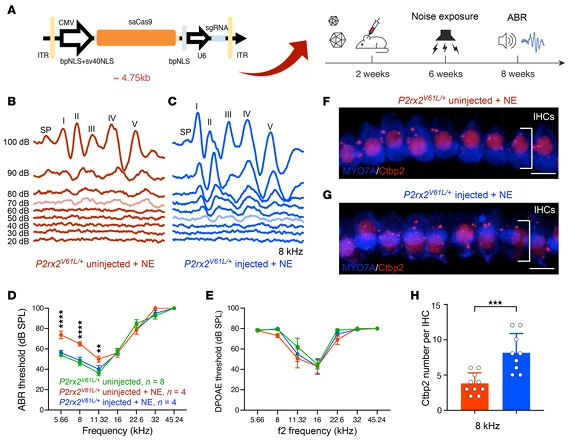
Figure 6. AAV2-SaCas9-sgRNA1 Injection Mitigates Increased Noise-Induced Hearing Loss (NIHL) Sensitivity in the DFNA41 Mouse Model (P2rx2^V61L/+)
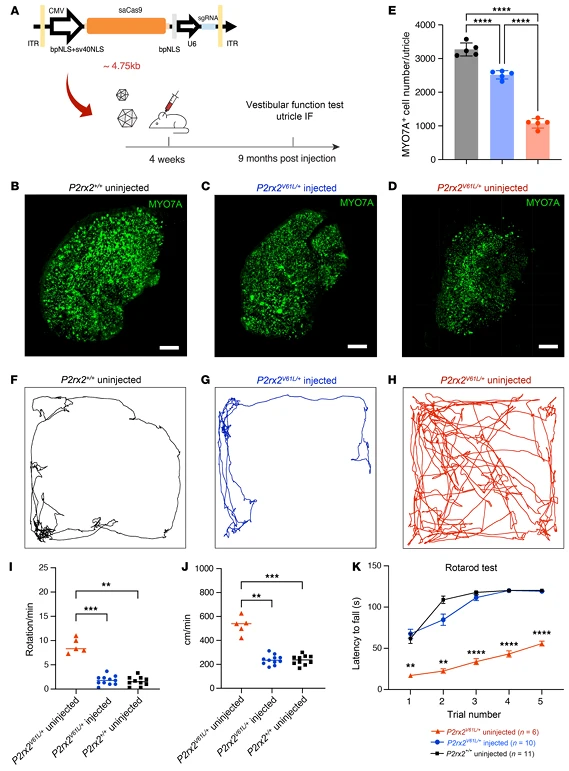
Figure 7. Adult Injection of V2-SaCas9-sgRNA1 Restores Vestibular Hair Cells and Function in the DFNA41 Mouse Model (P2rx2^V61L/+)
5.Clinical Translation of Genome Editing Tools Targeting Human P2RX2 V60L
To advance therapeutic development toward clinical application, the research team screened genome editing tools targeting the human P2RX2 V60L mutation (c.178G>T) in patient-derived human induced pluripotent stem cells (hiPSCs):
- SaCas9-KKH/sgRNA1 achieved a mutant allele editing efficiency of 37.9% ± 2.3%, with 76% of indels causing frameshift mutations.
- In contrast, enosCas12f1/sgRNA2 (4.6% ± 2.2%) and Cas12j-8 (<3%) showed very low editing efficiencies.
These results establish SaCas9-KKH/sgRNA1 as the optimal tool. Importantly, this system did not edit the wild-type P2RX2 allele, and no indels were detected at 10 bioinformatically predicted off-target sites unrelated to auditory function.These findings provide critical preclinical safety and efficacy data to support further translational studies and IND application for genome editing–based therapies for DFNA41.
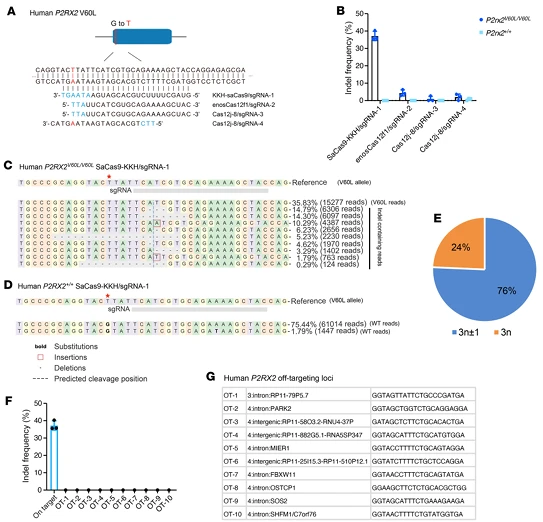
Figure 8. Allele-Specific Genome Editing of the P2RX2 V60L Allele in Patient-Derived hiPSCs Using a Compact CRISPR System
Summary and Outlook: From Animal Studies to Clinical Translation
This study systematically demonstrates that the AAV2-SaCas9-sgRNA1 editing system enables efficient and safe allele-specific genome editing in adult DFNA41 mouse models. The intervention successfully rescued auditory function, reduced noise sensitivity, and restored vestibular function, while also identifying an optimal genome editing tool for the human P2RX2 V60L mutation. Collectively, these results provide a comprehensive evidence chain from molecular correction to whole-organism phenotypic rescue, establishing a strong foundation for curative therapy for DFNA41.Nevertheless, several areas require further optimization:
- High-frequency hearing restoration: Adult mice exhibited complete loss of basal cochlear hair cells (responsible for high-frequency hearing). This may be due to mechanical injury from injection or early degeneration of high-frequency hair cells. Future strategies could include refined delivery techniques (to minimize basal turn damage) or combination with hair cell regeneration approaches.
- Editing efficiency: Current hair cell editing rates (~27%) may limit the protective range. Higher efficiencies could further expand therapeutic effects.
- Human vestibular function: The impact of DFNA41 on vestibular function in humans remains unclear, highlighting the need for clinical studies assessing vestibular deficits in patients.
Looking forward, with optimized delivery methods, preclinical safety validation (e.g., in non-human primates), and exploration of combined therapeutic strategies, this genome editing approach has the potential to become the first clinical therapy for DFNA41. It may also serve as a technical paradigm for gene editing–based treatment of other adult-onset, dominantly inherited hearing loss disorders.
Ubigene Gene Knockout (KO) Cell Line Services
Ubigene has successfully generated 8,000+ gene knockout (KO) cell lines, with special pricing starting from $990 and delivery in as fast as 1-2 weeks. You can search our cell line bank to find the cell line you need, and if it is not available, we also offer customized gene knockout services. Leveraging our proprietary CRISPR technology, we have achieved high-efficiency gene knockout in over 300 cell types, with editing efficiency 10–20 times higher than traditional methods. All knockout cell lines undergo dual verification by PCR and Sanger sequencing, ensuring precise genome editing and reliable experimental data.
Contact us to learn more >>>Reference
Wei W, Zhu W, Silver S, Armstrong AM, Robbins FS, Rameshbabu AP, Walz K, Quan Y, Du W, Kim Y, Indzhykulian AA, Shu Y, Liu XZ, Chen ZY. Single-dose genome editing therapy rescues auditory and vestibular functions in adult mice with DFNA41 deafness. J Clin Invest. 2025 Aug 14;135(20):e187872. doi: 10.1172/JCI187872. PMID: 41090360; PMCID: PMC12520686.
 Promotions
Promotions 








