Literature Review: Discovering Drug-Resistant Mutations with an Activation-Based Screening Platform

Literature Review: Discovering Drug-Resistant Mutations with an Activation-Based Screening Platform

Drug resistance is almost inevitable during targeted cancer therapy. Among the underlying causes, genetic mutations are predominant, yet identifying resistance-conferring variants in drug-treated patients is time- and cost-intensive. A recent Cell Reports article—“Accelerated drug-resistant variant discovery with an enhanced, scalable mutagenic base editor platform”—describes a new screening platform that couples a cytosine base editor (BE4) with a conditionally activated AID–dCas9 system (activation-induced cytidine deaminase fused to nuclease-dead Cas9). The engineered editor exhibits high efficiency, a broad editing window, and marked heterogeneity, enabling rapid discovery, classification, and functional annotation of drug-resistant mutations and providing a scalable tool for dissecting both cis- and trans-acting variants.
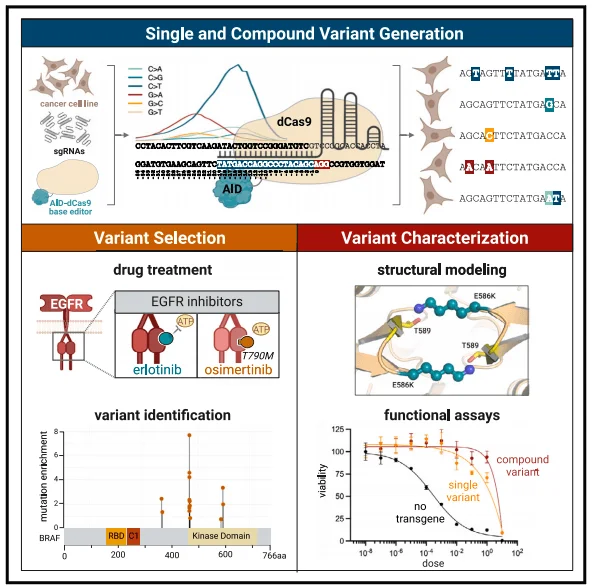
Figure 1. Overview of the study design
AID-dCas9 System: Innovations and Advantages
Efficient and Diversified Genome Editing
The AID-dCas9 system was engineered through the following modifications based on the conventional base editor BE4 architecture: Firstly, rAPOBEC1 was replaced with a hyperactive AID variant (AID*delta) that is directly fused as a cytidine deaminase. Compared to APOBEC1, AID exhibits a broader editing window while maintaining high editing efficiency and predominant C-to-T specificity. Secondly, by removing the uracil DNA glycosylase inhibitor (UGI) domain tethered in tandem with BE4, the system prevents uracil excision and promotes error-prone DNA repair pathways, enhancing mutation diversity. In the absence of the UGI domain, the editing specificity of AID-Cas9n extends beyond C>T to include C>A and C>G edits. Finally, dCas9 was employed instead of Cas9n to avoid interference from nickase activity. Compared to Cas9n, dCas9 effectively prevents DNA breaks, reduces indels, and generates more diverse editing types. Although overall editing efficiency is lower than that of AID-Cas9n, a significant increase in G>A, G>C, and G>T edits was observed near the 5' end of the sgRNA target sequence (positions -28 to -19 bp relative to the PAM site).
Additionally, beyond inducing single-base mutations, the AID-dCas9 system can generate compound variants through a single editing event and, when combined with appropriate sequencing methods, identify these diverse mutations while accommodating the identification of both cis and trans effects.
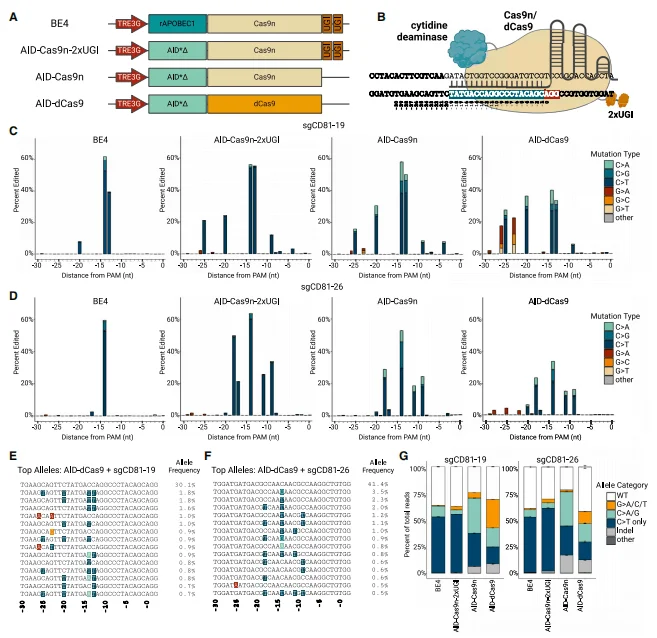
Figure 2. Optimization and characterization of the highly mutagenic AID–dCas9 base editor
Targeted Editing with AID-dCas9 System
To test the targeted editing ability of the AID-dCas9 system, 21 sgRNAs were designed to densely cover a 140 bp region of the fifth exon of CD81 (CD81ex5). Individual sgRNAs were delivered into PC-9 cells via lentiviral vectors, followed by 6-day doxycycline (DOX) treatment and sequencing of the edited products to analyze editing efficiency. Additionally, to control the exposure time and duration of each base editor in the genome, the single base editor was constructed downstream of a DOX-inducible promoter within a piggyBac transposon.
The results indicated that, while most C-to-N edits were concentrated upstream of the PAM site from -21 to -12 bp, G-to-N edits were enriched upstream of the PAM site from -28 to -19 bp—greatly and significantly widening the editing window compared with BE4. Approximately 45% of all nucleotides in the CD81 amplicon and around 80% of GC-rich nucleotides were editable by AID-dCas9, demonstrating its extensive editing range.
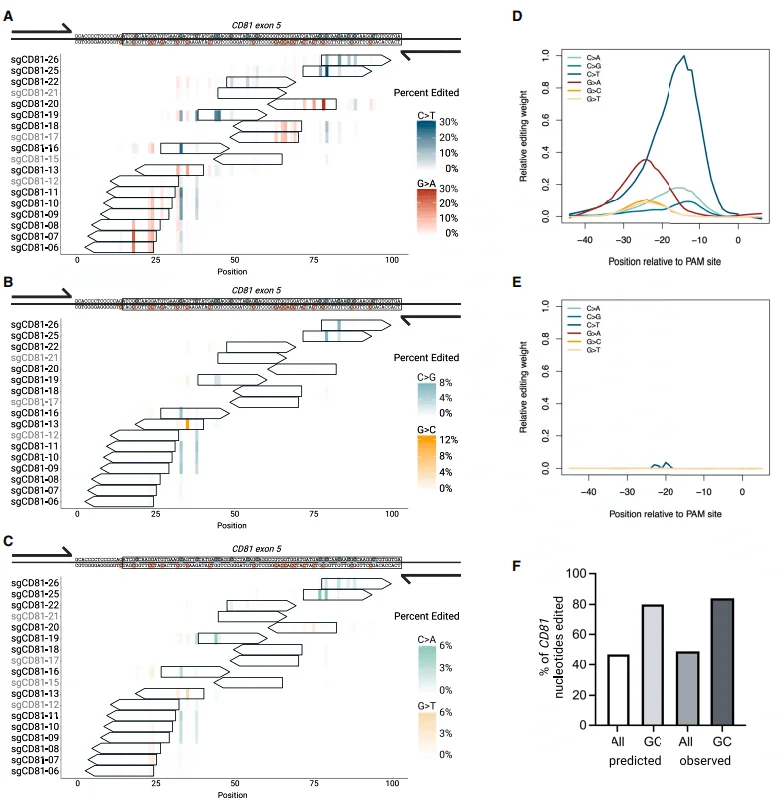
Figure 3. Targeted AID-dCas9 coupled with tiled sgRNAs achieves saturating mutagenesis
Applying the AID–dCas9 System to Drug-Resistance Screening
After validating mutational density, the authors assessed the platform in a therapeutic context. A pooled sgRNA library tiling all NGG PAM sites across EGFR and its downstream effector BRAF was constructed, alongside libraries for ESR1 (non-MAPK control) and 50 non-targeting guides. PC-9 cells stably expressing DOX-inducible AID–dCas9 were transduced, subjected to DOX, and treated with erlotinib, osimertinib, or vehicle. Surviving cells were harvested for sgRNA enrichment and targeted sequencing.
Both erlotinib and osimertinib were screened at both high and low doses: low doses were used to enrich a broader range of drug-resistant mutations to avoid missing potential mutations, while high doses enriched for more potent and robust resistance variants. Additionally, researchers tested whether the number of days of DOX treatment led to significant differences in sgRNA enrichment. The results showed significant enrichment of sgRNAs associated with drug resistance: sgRNAs enriched in four EGFR regions and two BRAF regions were highly significant. Specifically, significantly enriched sgRNAs overlapping with EGFR region 2 (corresponding to the T790 mutation site) were commonly present in all three groups except the high-dose osimertinib group, while sgRNAs in BRAF region 1 (corresponding to the G469 mutation site) were enriched in all three groups except the high-dose erlotinib group.
Targeted genomic sequencing of cells collected from the erlotinib and low-dose osimertinib groups revealed mutations at the T790M site, with the T790M mutation accounting for a higher proportion relative to other alleles at this site. Meanwhile, in the low-dose erlotinib and osimertinib groups, T790M and L792F mutations coexisted in cis. Sequencing of BRAF region 1 also revealed that both BRAF G469A and T470I not only generated single-point mutations but also exhibited cis-coexistence between the two. Therefore, the AID-dCas9 system can successfully generate known clinical drug-resistant mutations, validating the reliability of this platform.
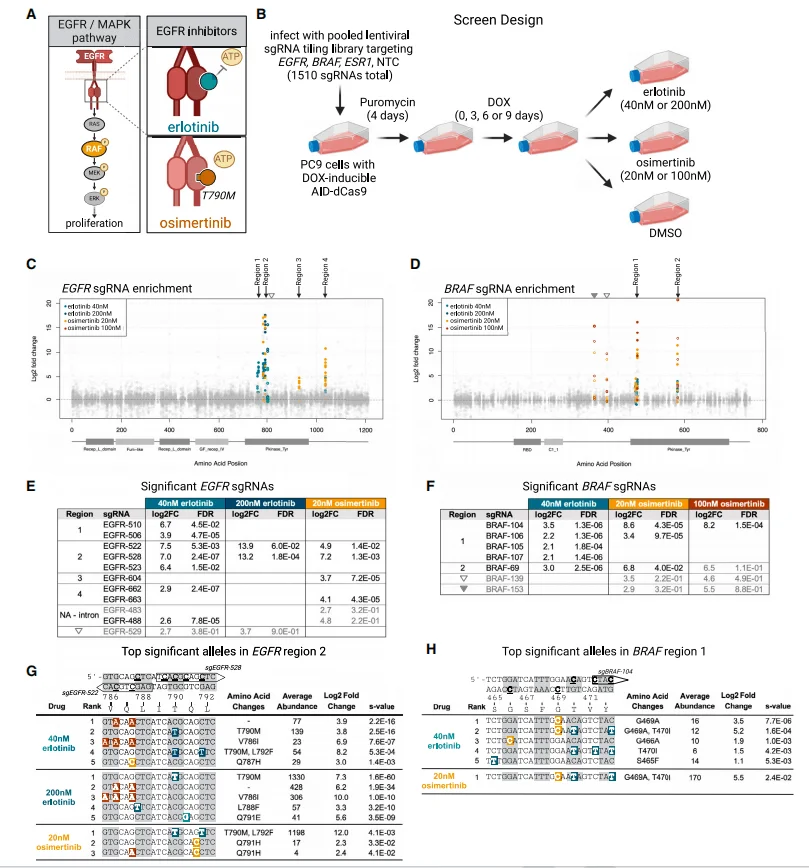
Figure 4. Pooled base-editing screen for EGFR-inhibitor resistance.
To conduct granular statistics on erlotinib/osimertinib-resistant variants edited by the AID-dCas9 system, researchers combined single-cell sequencing with secondary screening of the previously identified sgRNAs, utilizing the Tapestri platform for genotyping to assess genomic editing outcomes. One mixed sgRNA library was designed to target six strong mutation regions (EGFR1-4 and BRAF1-2), four weak mutation regions, one intronic junction region (to demonstrate that resistance arises from coding region mutations), and one negative control region. After constructing the library into vectors, it was introduced into AID-dCas9-PC-9 cells. Cells expressing the base editor were transfected with the sub-library, followed by puromycin treatment, DOX induction, and finally administration of erlotinib or osimertinib at IC95 doses. Single-cell sequencing was then performed to detect the sites targeted by each sgRNA. Each sequencing sample included approximately 4,000 cells, with each amplicon yielding around 20,000-200,000 reads.
For significantly enriched variants in EGFR, amino acid mutations occurred primarily in the erlotinib group and clustered in the EGFR kinase domain; among these, T790M and the adjacent Q791E mutation were particularly prominent, while L792F and other T790-proximal mutations also showed high enrichment. Single-cell data were aggregated and subjected to pseudo-bulk analysis to determine the alleles of these mutations: in the erlotinib group, the top three significantly enriched alleles in EGFR (ranked by S value) all contained the T790M mutation (T790M, T790M/Q791E, and T790M/L792F), indicating that this mutation is a major driver of resistance. In the osimertinib group, only one significantly enriched allele (T790M/L792F) was identified, which is consistent with previous research showing that “additional mutations such as L792F confer resistance to osimertinib only when present in cis with T790M.”
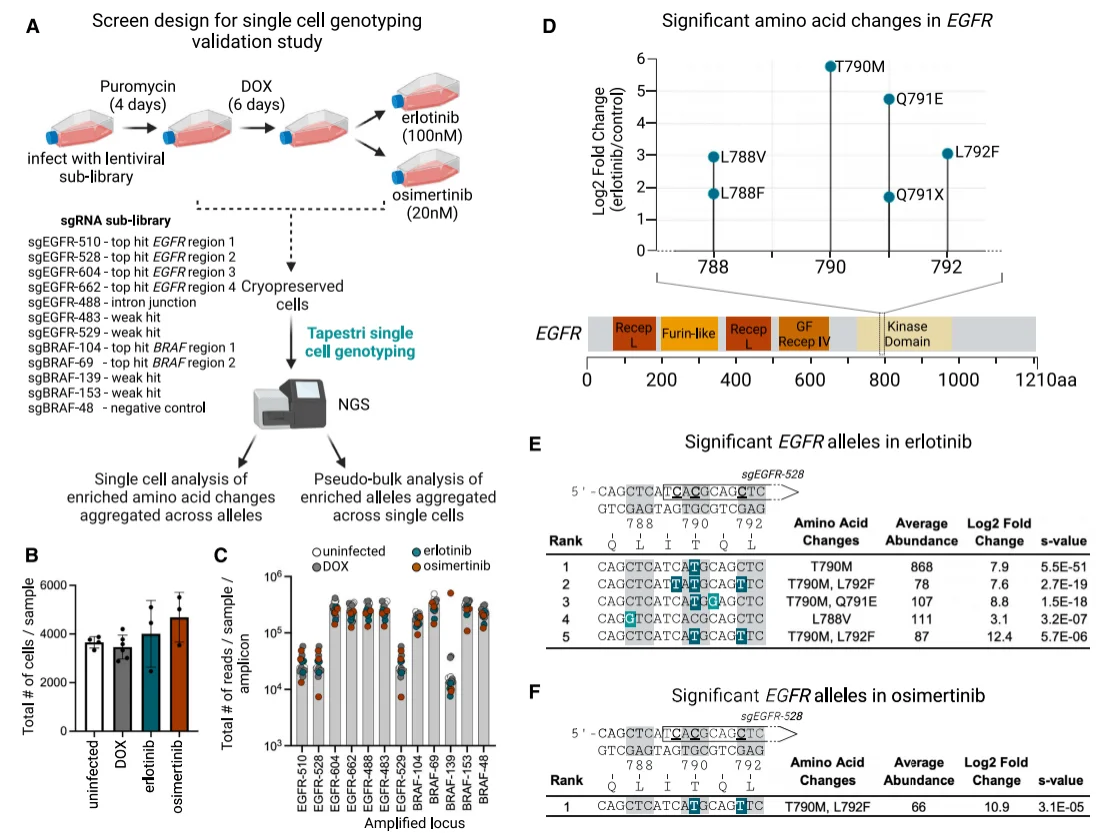
Figure 5. Single-cell genotyping of EGFR mutations conferring resistance.
Analyzing single amino acid mutations in BRAF revealed three regions with enriched mutations in the osimertinib group, including the most important and widely studied G469 locus—both single and compound mutations at this locus were significantly enriched. Following treatment with both osimertinib and erlotinib, G469A alleles coexisted in cis with T470I or T470K alleles; notably, the G469/T470 compound mutations represent a novel variant type identified through the AID-dCas9 system screening.
Another set of significantly enriched mutations spanning the E586-D594 sequence in the BRAF kinase domain includes the class II BRAF mutant E586K and class III mutant D594N; similarly, the role of the single or compound mutations of E586K/D594N in resistance to EGFR small-molecule inhibitors has not been previously reported. Additionally, two alleles encoding the A366P mutation were significantly enriched in the osimertinib group.
Since these resistance mutations at the aforementioned loci have not been extensively studied, the authors performed protein structure modeling for these mutations to explain the resistance mechanisms by correlating phenotypes with target structures.
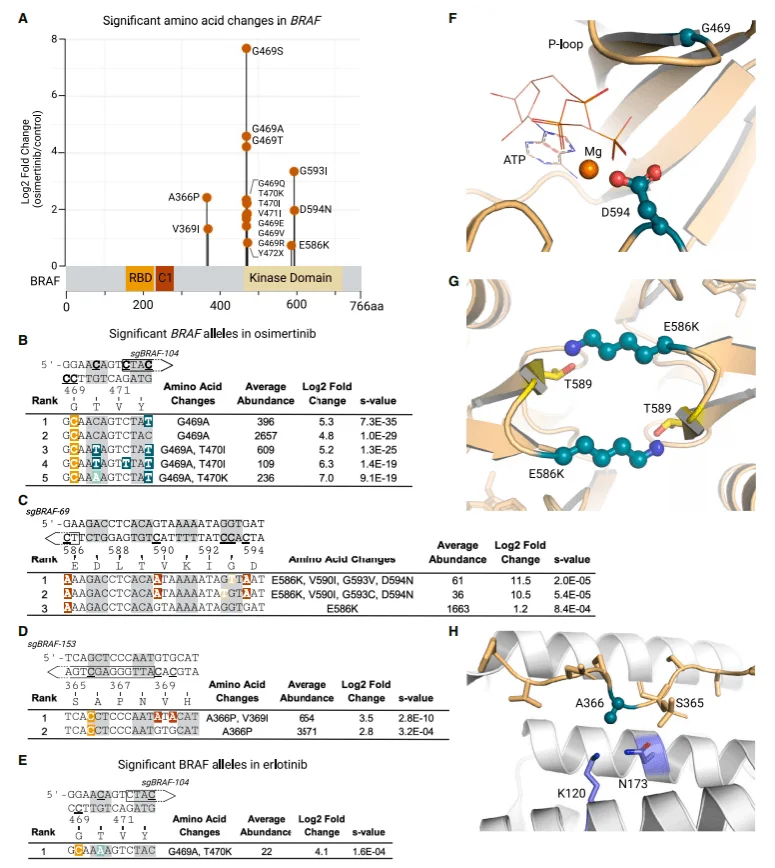
Figure 6. Single-cell genotyping of BRAF variants enriched post-EGFR inhibition
Following the identification of potential drug-resistant mutation sites, researchers validated these mutations (particularly BRAF-related ones) through complementary overexpression experiments: A series of mutations were overexpressed in a DOX-inducible PC-9 cell model, with EGFR mutations containing the activating exon 19 deletion (del746-750) serving as a positive control. Results showed that both the T790M single point mutation (in the erlotinib group) and the T790M/L792F compound mutation (in the erlotinib and osimertinib groups) synergistically enhanced resistance in combination with the exon 19 deletion mutation.
Following BRAF overexpression, significant enhancement of resistance was observed under different drug types or doses: While the BRAF G469A single point mutation conferred cellular resistance to erlotinib and osimertinib, its two compound mutations did not enhance this effect. In contrast, the presence of the E586K/D594N/G593V/V590I compound mutation significantly increased resistance across doses of erlotinib and osimertinib, surpassing the resistance induced by the E586K single point mutation. Similarly, the BRAF A366P single point mutation also enhanced cellular resistance to erlotinib and osimertinib to some extent.
Western blot analysis of MAPK pathway activation markers (including phosphorylated EGFR, phosphorylated ERK, and phosphorylated MEK) was conducted on cell lines carrying DOX-inducible mutant EGFR or BRAF transgenes, under conditions with or without DOX, erlotinib, and osimertinib. Additionally, western blot results showed that under EGFR inhibitor (erlotinib/osimertinib) treatment, pMEK/pERK expression (downstream regulators in the EGFR pathway) was significantly inhibited in the control group, indicating pathway inactivation; in contrast, sustained high expression of pMEK/pERK in the BRAF mutant group indicated continuous pathway activation.
This indicates that BRAF mutants can sustain the downstream signaling of the EGFR/MAPK pathway independently of EGFR, thereby enhancing resistance and enabling cell survival.
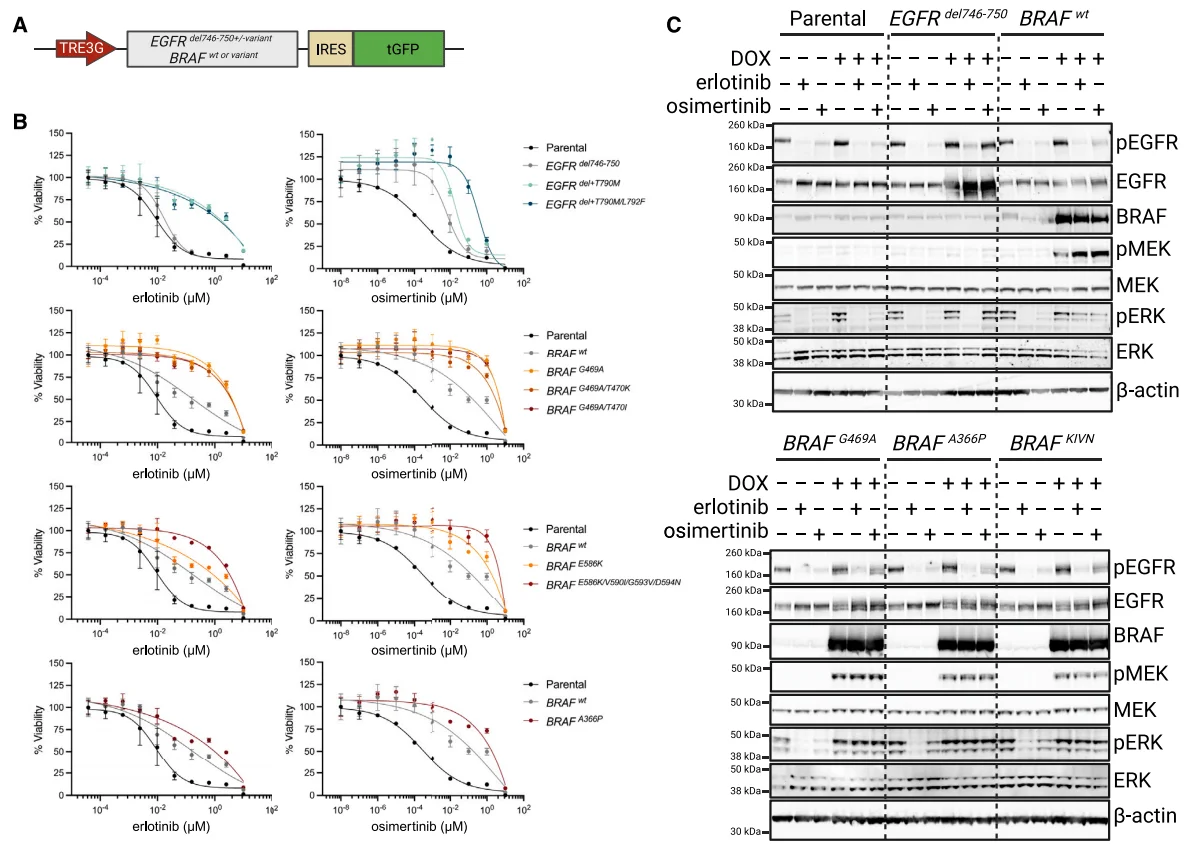
Figure 7 Overexpression of DOX-inducible EGFR and BRAF mutant genes recapitulates the EGFR inhibitor resistance phenotype
Summary
This study introduces an AID–dCas9 base-editing platform that achieves broad and heterogeneous mutagenesis through three engineering steps: (1) hyperactive AID, (2) UGI deletion, and (3) dCas9 substitution. The system enables saturation mutagenesis of clinically relevant loci and rapid identification of known and novel resistance alleles. The tiered screening strategy—tiled library → pooled selection → single-cell genotyping—minimizes false negatives and is readily adaptable to other genes, pathways, or therapeutic modalities.
The overall experimental design of this study is well-structured and progressive: it successfully validates editing diversity on the model gene CD81 before extending this validation to the context of actual drug screening to assess the ability of the AID-dCas9 system to generate drug-resistant mutations. A stratified strategy is employed in the screening process, initially using a tiled design approach to create an sgRNA library targeting relevant signaling pathways for preliminary screening, identifying sgRNA-enriched regions to enhance efficiency and narrow the scope. This is followed by a focused screening that covers these mutation-enriched regions and integrates single-cell genotyping for high-precision analysis, ultimately identifying potential drug-resistant mutation sites. Additionally, the design of the sgRNA library, selection of controls, stratified drug dosage screening, and optimization of the editing-screening time window are all quite clever. By integrating these with high-coverage sequencing, the design effectively avoids the oversight of weak mutations. This methodology is not only applicable to research on tumor drug resistance but can also be referenced in exploring genes or pathways related to disease phenotypes, discovering targets for enhancing cell therapy or immunotherapy, identifying relevant factors that promote cellular fate transitions, and searching for factors that enhance viral infectivity or host defense factors against viruses, among other research areas.
Whether you are exploring unknown mechanisms, screening drug targets, tackling resistance challenges, or optimizing immunotherapy strategies, Ubigene Biosciences is always your trusted research partner. We offer comprehensive CRISPR functional screening solutions that include customized solution design (tailored screening strategies based on research questions),efficient and stable experimental execution (ensuring data quality through a mature platform), seamless full-process integration (covering all aspects of CRISPR library screening), and professional data interpretation support (helping you extract biological insights from massive amounts of data). If you are looking for one-stop, high-quality functional screening services, feel free to contact us and embark on a new chapter in precision research!
Reference
Dorighi KM, Zhu A, Fortin JP, Hung-Hao Lo J, Sudhamsu J, Wendorff TJ, Durinck S, Callow M, Foster SA, Haley B. Accelerated drug-resistant variant discovery with an enhanced, scalable mutagenic base editor platform. Cell Rep. 2024 Jun 25;43(6): 114313. doi:
 Promotions
Promotions 








