EMBO J | FOXP1 Regulates ATR Activation and Replication Stress Response via Antagonistic Phosphorylation and O-GlcNAcylation


The ATR signaling pathway constitutes a central safeguard against replication stress. Dysregulation of ATR activation leads to genomic instability and contributes to the development of cancer and other diseases. FOXP1, a member of the Forkhead box (FOX) family of transcription factors, has traditionally been characterized as a regulator of gene transcription.In a study published in The EMBO Journal, research teams led by Professor Xingzhi Xu from the School of Medicine, Shenzhen University, and Professor Peng Gong from Shenzhen University General Hospital report a previously unrecognized, non-transcriptional role of FOXP1 in the DNA damage response. The study demonstrates that FOXP1 functions as a scaffold protein, directly interacting with the RPA-ssDNA complex and the ATR-ATRIP complex to facilitate ATR recruitment and activation at stalled replication forks.
Importantly, the activity of FOXP1 is tightly regulated by a dynamic interplay between O-GlcNAcylation and phosphorylation. O-GlcNAc transferase (OGT)-mediated O-GlcNAcylation of FOXP1 suppresses its interaction with ATR, thereby limiting ATR activation under basal conditions. Upon replication stress, CHK1-mediated phosphorylation of FOXP1 at serine 396 (S396) antagonizes O-GlcNAcylation at this site, forming a positive regulatory loop that promotes efficient ATR signaling.Pathogenic FOXP1 mutations identified in tumor tissues disrupt this regulatory mechanism, resulting in defective ATR activation and compromised replication fork stability. These findings establish FOXP1 as a critical modulator of the replication stress response and highlight its non-transcriptional function in maintaining genome integrity.
Ubigene provided FOXP1 S396A and S396D point-mutant HEK293 cell lines for this study, enabling mechanistic dissection of phosphorylation-dependent regulation of FOXP1. This support facilitated the identification of FOXP1 as a novel molecular regulator of ATR signaling and suggests new potential therapeutic targets for cancers associated with replication stress dysregulation.

Research Background
During DNA replication, cells are continuously challenged by both endogenous stresses—such as aberrant dNTP synthesis—and exogenous insults, including ultraviolet irradiation and chemotherapeutic agents. These stresses can lead to replication fork stalling, which, if not properly resolved, results in DNA breakage and genomic instability.The ATR-CHK1 pathway represents a central signaling network in the cellular response to replication stress. Activation of the ATR kinase requires its recruitment to RPA-coated single-stranded DNA (RPA-ssDNA) and subsequent stimulation by activator proteins such as TopBP1 and ETAA1. This process is precisely regulated by multiple post-translational modifications (PTMs), including phosphorylation and ubiquitination.
O-GlcNAcylation is another major PTM that frequently targets serine and threonine residues shared with phosphorylation sites. These two modifications often exhibit competitive or cooperative interactions, thereby fine-tuning protein function. However, the role of O-GlcNAcylation in ATR signaling and replication stress responses remains largely unexplored.FOXP1 is a multifunctional transcription factor whose haploinsufficiency has been associated with developmental delay and tumorigenesis. Notably, the clinical manifestations of FOXP1 deficiency show similarities to phenotypes arising from impaired ATR signaling. Despite this correlation, whether FOXP1 directly participates in the replication stress response, and the molecular mechanisms underlying its regulation, have remained unclear.
Research Objectives
This study aims to elucidate the functional role of FOXP1 in ATR activation and the replication stress response, to dissect the regulatory interplay between O-GlcNAcylation and phosphorylation of FOXP1, and to define the molecular mechanisms by which pathogenic FOXP1 mutations compromise replication stress signaling. These insights may identify FOXP1 as a potential therapeutic target in cancers characterized by dysregulated replication stress responses.
Research Methods
Cellular and Molecular Assays: HEK293T, H1975, and additional cell lines were employed for cellular and molecular analyses. Protein-protein interactions were examined using immunoprecipitation (IP), GST pull-down assays, and immunoblotting (IB). FOXP1 expression and mutational status were manipulated through siRNA-mediated knockdown and CRISPR-Cas9-based knock-in strategies. Post-translational modifications of FOXP1 were assessed using in vitro phosphorylation assays and O-GlcNAcylation detection approaches.
Functional Validation Assays: Replication fork stability was evaluated using DNA fiber assays. Cell cycle distribution and S-phase arrest were analyzed by flow cytometry. Cellular sensitivity to replication stress-inducing agents, including hydroxyurea (HU) and camptothecin (CPT), was assessed using cell viability assays. The localization of FOXP1 at replication forks was investigated through immunofluorescence microscopy, proximity ligation assays (PLA), and isolation of proteins on nascent DNA (iPOND).
Mutant Analysis: Pathogenic FOXP1 variants (including R465G and R514C) as well as post-translational modification-related mutants (such as S396A and S396D) were generated to determine their effects on protein interactions, ATR activation, and replication fork stability.

Figure 1. Working Model of FOXP1 Function in the Replication Stress Response
Research Workflow
Identification and Validation: ATR-interacting proteins were screened by immunoprecipitation followed by mass spectrometry (IP-MS), leading to the identification of FOXP1 as a candidate regulator. The interactions of FOXP1 with the ATR-ATRIP complex and the RPA-ssDNA complex were subsequently validated, and the enhancement of these interactions under replication stress conditions was examined.
Functional Characterization: FOXP1 was depleted to assess its impact on ATR activation, as measured by CHK1 phosphorylation at serine 345 (CHK1 S345). Replication fork stability and cellular sensitivity to replication stress were also evaluated, establishing FOXP1 as a positive regulator of the ATR signaling pathway.
Mechanistic Dissection of Post-Translational Modifications: OGT-mediated O-GlcNAcylation of FOXP1 was characterized, and its inhibitory effect on FOXP1-ATR interaction was determined. In parallel, CHK1-mediated phosphorylation of FOXP1 at serine 396 (S396) was identified and shown to antagonize O-GlcNAcylation, thereby promoting ATR activation under replication stress.
Functional Analysis of FOXP1 Mutants: Tumor-associated FOXP1 mutants were analyzed to determine their effects on DNA binding, protein-protein interactions, ATR activation, and replication fork stability, elucidating the molecular basis of their pathogenicity.
Integrated Mechanistic Model: An integrated molecular model of ATR activation regulated by dual post-translational modifications of FOXP1 was established, defining FOXP1 as a central regulator in the cellular replication stress response.
Key Findings
FOXP1 Promotes ATR Activation: FOXP1 directly interacts with the ATR-ATRIP complex, and this interaction is markedly enhanced under hydroxyurea (HU)-induced replication stress. Depletion of FOXP1 significantly attenuates ATR-dependent phosphorylation of CHK1 at serine 345 (CHK1 S345), compromises replication fork stability, and increases cellular sensitivity to HU. These results demonstrate that FOXP1 functions as a positive regulator of ATR activation and the replication stress response.
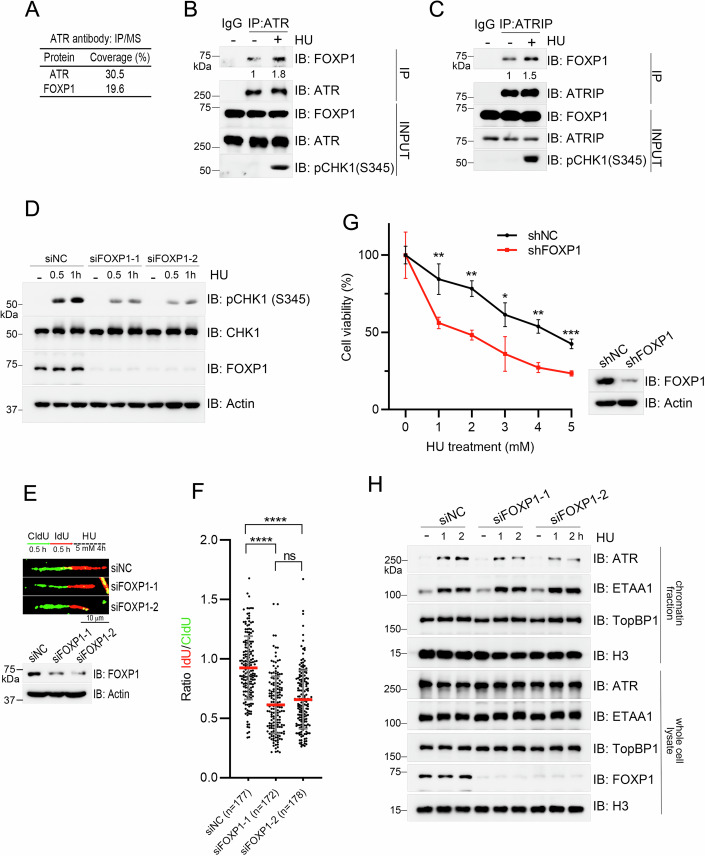
Figure 2. FOXP1 Promotes ATR Activation under Replication Stress
FOXP1 localizes to stalled replication forks in response to replication stress: FOXP1 directly binds single-stranded DNA (ssDNA) via its forkhead domain (amino acids 465-555) and exhibits higher affinity for RPA-coated ssDNA. FOXP1 also directly interacts with RPA70 and RPA32 and is recruited to stalled replication forks in an RPA-dependent manner under replication stress conditions.
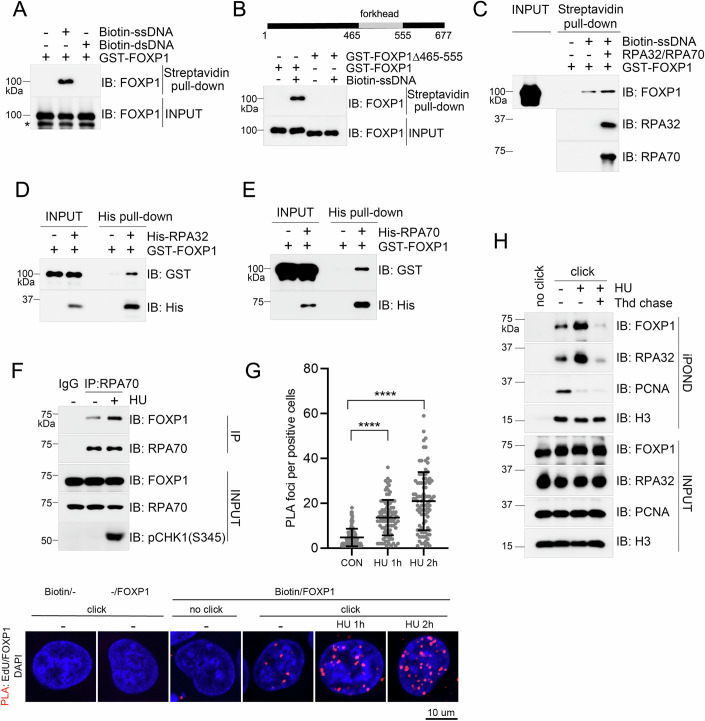
Figure 3. FOXP1 Associates with Stalled Replication Forks
O-GlcNAcylation suppresses the interaction between FOXP1 and ATR: O-GlcNAc transferase (OGT) directly catalyzes O-GlcNAcylation of FOXP1 through its 556-610 amino acid region. Under replication stress conditions, the O-GlcNAcylation level of FOXP1 is reduced. This modification negatively regulates the association between FOXP1 and ATR, thereby limiting ATR activation.
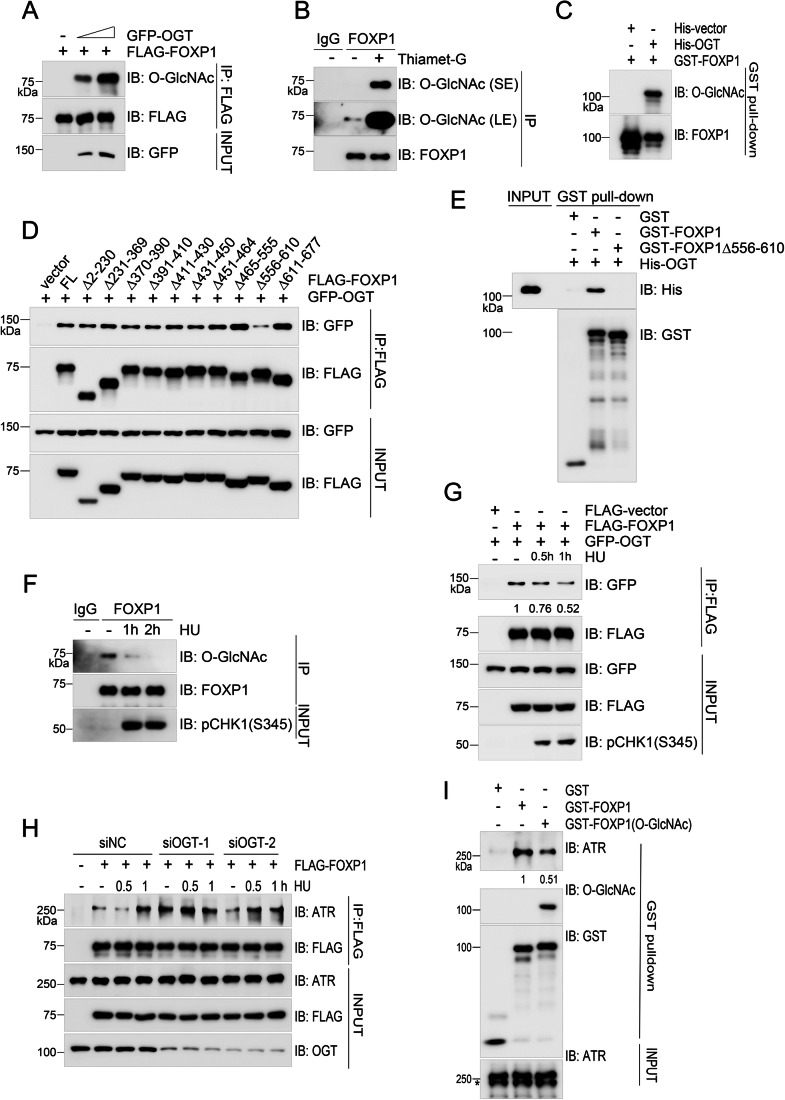
Figure 4. OGT-mediated O-GlcNAcylation of FOXP1 inhibits its interaction with ATR
CHK1-mediated phosphorylation antagonizes O-GlcNAcylation: CHK1 directly phosphorylates FOXP1 at the S396 residue. The phosphorylation-deficient mutant S396A exhibits increased O-GlcNAcylation, reduced interaction with ATR, and impaired ATR activation. In contrast, the phospho-mimetic mutant S396D shows decreased O-GlcNAcylation, enhanced ATR binding, and effectively rescues ATR activation defects.
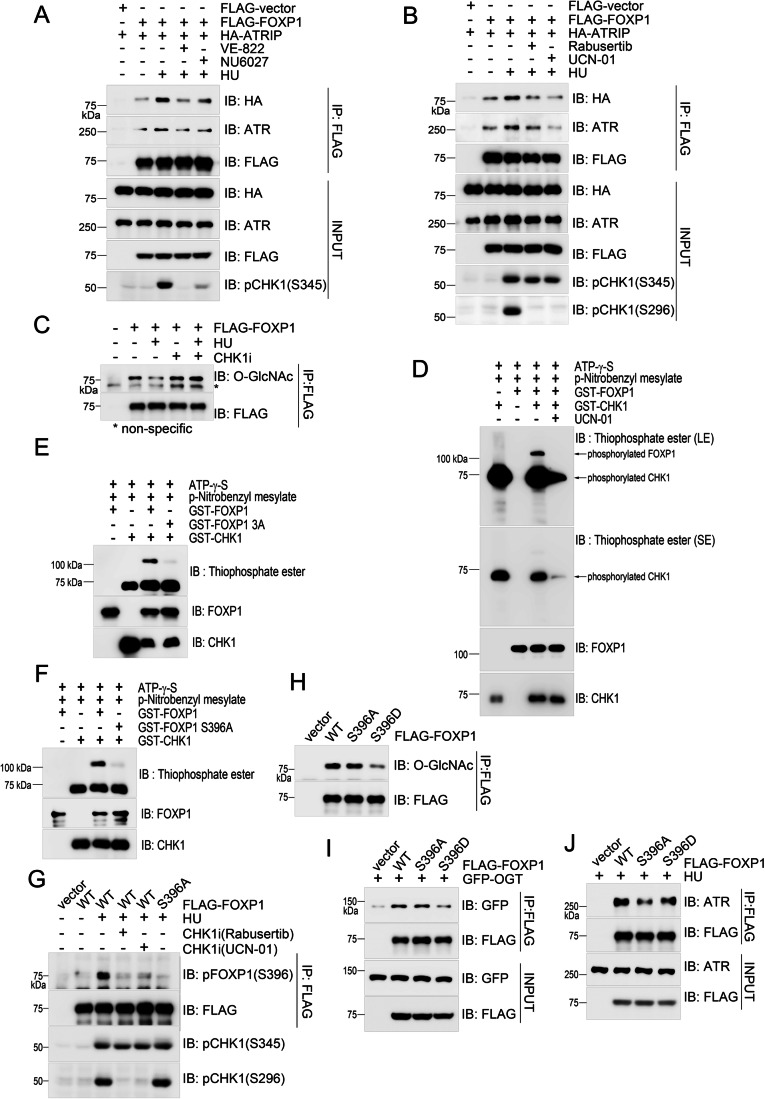
Figure 5. CHK1-mediated phosphorylation of FOXP1 at S396 antagonizes its O-GlcNAcylation
Functional defects of FOXP1 mutants: Tumor-associated FOXP1 mutants (e.g., R465G, R514C) exhibit reduced binding to ssDNA and RPA, increased O-GlcNAcylation levels, and weakened interaction with ATR. These alterations ultimately result in impaired ATR activation and decreased replication fork stability.
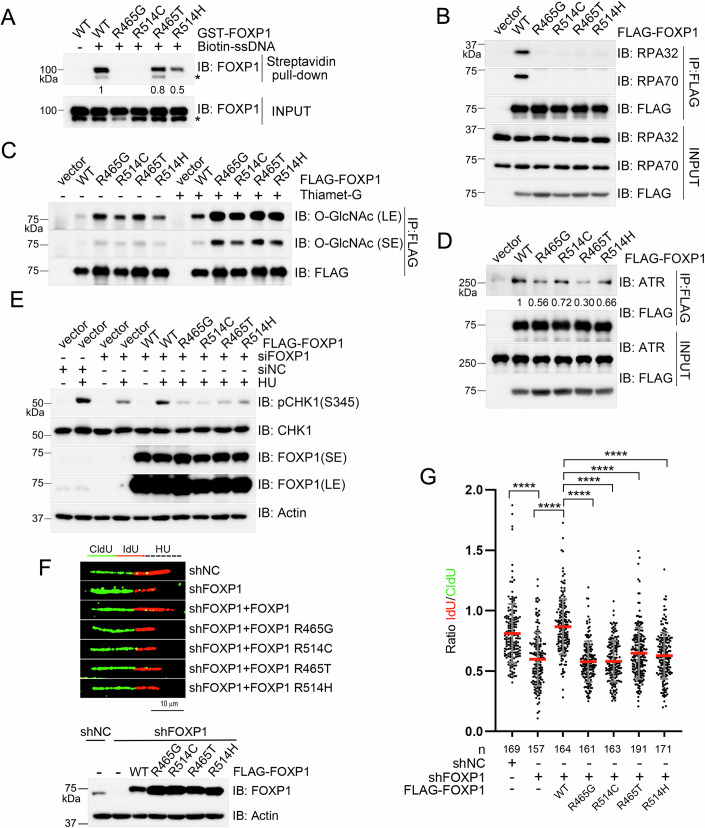
Figure 6. Pathogenic FOXP1 mutations impair its function under replication stress
Significance and Innovation
Theoretical Innovation: This study uncovers a non-transcriptional function of FOXP1 for the first time, demonstrating its role as a scaffold protein mediating ATR recruitment and activation, thereby expanding the known biological functions of FOXP1. It also reveals the critical antagonistic interplay between O-GlcNAcylation and phosphorylation in regulating the ATR pathway, enriching the post-translational modification (PTM) network governing the replication stress response.
Mechanistic Breakthrough: Elucidate a positive regulatory loop—“CHK1 → FOXP1 (phosphorylation) → OGT → FOXP1 (O-GlcNAcylation) → ATR”—providing new insights into the precise control of ATR activation under replication stress.
Clinical Value: Tumor-associated FOXP1 mutants disrupt the replication stress response, promoting oncogenesis, and may serve as molecular biomarkers for tumor stratification. As a key regulator of ATR activation, FOXP1 represents a potential target for combinational cancer therapy, particularly with replication stress-inducing chemotherapeutics.
Summary
This study reveals, for the first time, the non-transcriptional role of the transcription factor FOXP1 in the replication stress response, functioning as a scaffold protein to mediate ATR recruitment and activation. FOXP1 activity is regulated by dual post-translational modifications: OGT-mediated O-GlcNAcylation inhibits its interaction with ATR, whereas CHK1-mediated phosphorylation at S396 under replication stress antagonizes this modification, enhancing FOXP1 recruitment to ATR and ensuring replication fork stability. Tumor-associated pathogenic FOXP1 mutations disrupt DNA binding, protein interactions, and PTM balance, leading to defective ATR activation, genomic instability, and tumorigenesis. This work not only enriches the regulatory network of the ATR signaling pathway but also provides critical mechanistic insights and potential therapeutic targets for FOXP1-related cancers.
Support provided by Ubigene
For this study, custom FOXP1 S396A and S396D point-mutant HEK293 cell lines provided by Ubigene served as critical experimental models to dissect the molecular mechanisms by which pathogenic FOXP1 mutations regulate the replication stress response.Leveraging years of expertise in gene editing, Ubigene has successfully completed over 13,000 gene-editing projects and established a mature and reliable technical service platform. For point mutation cell line construction, Ubigene offers a one-stop solution encompassing four technical approaches: RNP delivery, base editors, prime editors, and plasmid-based methods. These flexible strategies can accommodate various mutation scenarios with efficiency and ease.
With Ubigene's proprietary EZ-HRex™ technology, the highest HDR genotype frequency can reach up to 84%, providing researchers with highly efficient and precise gene-editing tools.
Reference
ZhuX,GaoC,PengB,XueJ,XiaD,YangL,ZhangJ,GaoX,HuY,LinS,GongP,XuX.FOXP1phosphorylationantagonizesitsO-GlcNAcylationinregulatingATRactivationinresponsetoreplicationstress.EMBOJ.2025Jan;44(2):457-483.doi:10.1038/s44318-024-00323-x.Epub2024Dec2.PMID:39623140;PMCID:PMC11729909.
 Promotions
Promotions 








