Expert Insights | Tips for GL261 Cell Culture and Gene Editing


Expert Insights | Tips for GL261 Cell Culture and Gene Editing
As a classic glioma model derived from C57BL/6 mice, GL261 cells have become a gold standard for studying glioblastoma (GBM) immunotherapy, tumor microenvironment regulation, radiochemotherapy sensitivity, and combination treatment strategies due to their clear immunogenicity and the unique advantage of being able to model in immunocompetent mice.
Whether you're delving into the PD-1/PD-L1 pathway mechanism, assessing the efficacy of CAR-T/NK cell therapies, or building a CRISPR/Cas9 functional screening platform, GL261 can help you precisely analyze brain tumor immune responses and drive efficient research breakthroughs! Today, Ubigene will unlock the secrets of using GL261—sharing practical cell culture techniques and high-efficiency experimental strategies to help you achieve high-quality results and steady progress in your glioblastoma research.
Order GL261 cell now>>>Cell Information
Cell Name: GL261 (Mouse Glioblastoma Cell Line)
Cell Morphology: Epithelial - like, adherent
Cell Culture Conditions: 90% DMEM + 10% FBS
Gas Phase: Air 95%; CO2, 5%
Temperature: 37°C
Media Change Frequency: Every 2-3 days
Passage Ratio: 1:2 - 1:3
Normal Cell Growth Image: Cells adhere well, with clear outlines and neat edges. During the culture process, there are no large amounts of debris or cell secretions between the cells.
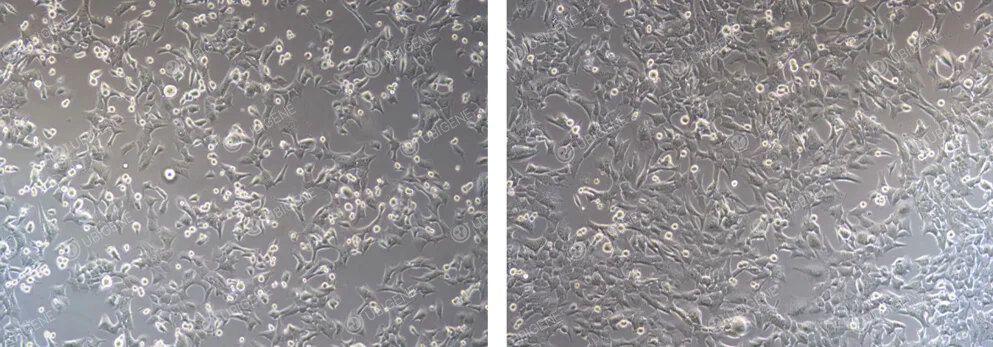
Figure 1. Normal GL261 Cell Growth
Abnormal Cell Growth Image: There are few adherent cells with indistinct cell outlines. Abundant cell debris and extracellular matrix are present in the culture.
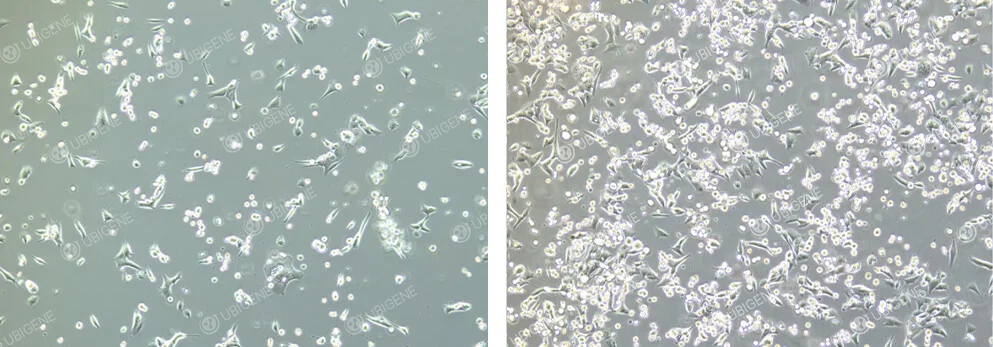
Figure 2. Poor GL261 Cell Growth
Cell Thawing
1)Preparation: Preheat the water bath to 37℃ and warm the complete culture medium.
2)Aliquot 7 mL of the preheated complete culture medium into 15 mL centrifuge tubes in a clean bench.
3)Take the cells from dry ice, hold the lid with tweezers, and place the tube into a 37℃ water bath. Rapidly shaking around 1 minute until fully thawed (water should not cover the lid).
4)Using a pipette, transfer the cell suspension to the 15 mL centrifuge tube prepared in step 2. Cap the tube and centrifuge at 1100 rpm for 4 minutes at room temperature.
5)Discard the supernatant in the clean bench. Resuspend the cell pellet in 1 mL of complete culture medium to create a single-cell suspension. Transfer the resuspended cells to a T25 cm² culture flask (or a 6 cm culture dish) containing 4 mL of complete culture medium. Label with cell name, resuscitation date, and passage number. Incubate at 37℃ with 5% CO2.
6)Observe the cell condition after 24 hours.
Cell Passaging
1)Passage cells when they reach 80-90% confluence. In clean bench, remove the culture medium from the culture flask and discard it into a waste container. Wash the cells 1-2 times with 1×PBS (approximately 2-3 mL for a T25 cm² flask, and 4-5 mL for a T75 cm² flask) to remove residual medium and serum (serum contains trypsin inhibitors).
2)Add an appropriate volume of trypsin solution (refer to Table 1 below for specific volumes). Gently shake the flask to ensure the trypsin completely covers the cells. Place the flask in the incubator for 1-2 minutes. Observe under a microscope, once most cells appeared rounded and detached from the surface upon gently shake and tap the culture flask, immediately stop digestion.
| Size of culture plates/flasks | Trypsin volume |
|---|---|
| 6-well plate | 0.5 mL |
| T25 | 1 mL |
| T75 | 2-3 mL |
| T175 | 3-4 mL |
3)Add double trypsin volume of complete culture medium to neutralize the trypsin and gently resuspend the cells by pipetting several times to ensure all cells are detached. Note: resuspend cells gently to minimize or avoid bubbles.
4)Transfer the cell suspension to a 50 mL centrifuge a tube using a 10 mL pipette. Cells from the same batch can be pooled together. Wash the flask with an appropriate volume of PBS to recover any remaining cells and add to the 50 mL tube. Cap the tube and label it appropriately.
5)Centrifuge at 1100 rpm for 4 minutes at room temperature. After centrifugation, discard the supernatant and resuspend the cell pellet in 2 mL of complete culture medium.
6)Cells should be passaged at a defined seeding ratio. For the first passage, a 1:2 passage ratio is recommended. If the cells reach full confluency within two days, the passage ratio can be increased in subsequent passages.
Cell Cryopreservation
1)Transfer all the cell suspension from the culture flask to a centrifuge tube.
2)Centrifuge at 1100 rpm for 4 minutes. Discard the supernatant, add an appropriate amount of 4℃ pre-cooled cryopreservation solution to resuspend the cells, then take 20μL of the cell suspension for cell counting, and adjust the cell density to 5×10^6 to 1×10^7 cells/mL.
3)Aliquot 1 mL into each cryogenic tube, place the tubes in a controlled-rate freezing container, then transfer to a -80℃ freezer.
4)Stay overnight, transfer the cryovials to liquid nitrogen for long-term storage
Suggestions for Handling Excessive Cell Debris, Secretions, or Cell Death During Culture or Post Drug Screening
1)If there are too many dead cells during cultivation, change the medium frequently (every day or every other day) and use high-quality fetal bovine serum.
2)If there are excessive cellular secretions or cell fragments during cultivation, first digest the cells, then wash the cell pellet with PBS. Resuspend the cells and transfer the cell suspension to a 5ml EP tube for centrifugation (centrifuge at 1100 rpm for 4 minutes). After centrifugation, carefully remove as much PBS as possible, this step can be repeated twice. Based on the amount of cells collected, seed them into an appropriate flask or dish.
Suggestions for Improving Low Cell Viability After Thawing
GL261 cells are susceptible to damage during cryopreservation. To improve cell viability upon thawing, a higher cell density is recommended during freezing, generally 1-2×10^6 cells/mL. Upon thawing, cells should initially be resuspended in 1-2 six-well plates to allow recovery before transferring to a larger culture flask.
How to Improve Monoclonal Formation Rate?
1)Cell viability should be >90% before plating for colony formation.
2)The final cell count after dilution should ideally be 1-2×10^6 cells/ml.
3)Use high - quality Australian fetal bovine serum when plating cells for colony formation.
4)Wash cell clumps twice with PBS before plating.
5)Conduct a preliminary experiment to determine the optimal cell plating dilution to avoid a low monoclonal ratio.
6)Ensure even cell distribution when seeding cells into 96-well plates.
Suggestions for Cell Transfection
1)Cell viability should be >90%, with cells in good condition, clear outlines, and neat edges.
2)Handle cells gently during transfection.
3)When using electroporation, select appropriate parameters to avoid cell death.
4)For viral transfection, a preliminary experiment is recommended to determine the optimal MOI (multiplicity of infection) and the best drug screening concentration; ensure the appropriate viral titer; add Polybrene before transfection; perform the infection in a 12-well plate.
Notes:
1)For cryopreservation, GL261 cells should have a viability >90%, with 5×10^6-1×10^7cells/ml per vial.
2)GL261 cells produce cellular secretions and debris during growth. It is recommended to wash the cell pellet with PBS every other passage to reduce the impact of these secretions and debris on cell proliferation and condition.
3)GL261 cell growth is density-dependent. The passage ratio should not be too high; a 1:2 to 1:3 ratio is recommended.
4)GL261 cells are sensitive to mechanical forces. Gently pipette the cells during routine culture to avoid excessive force, which can lead to cell differentiation and death.
5)Maintain appropriate cell density during culture and passage.
6)GL261 cells are sensitive to the quality of fetal bovine serum (FBS). High-quality Australian FBS is recommended.
Image of Cells After Electroporation
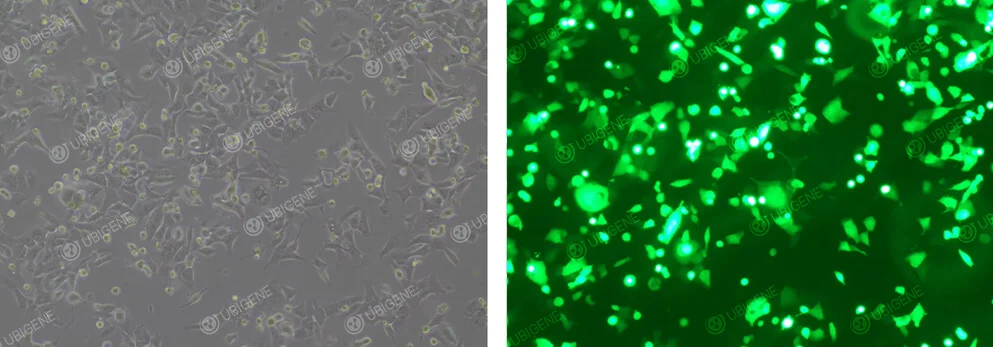
Figure 3. GL261 Cells After Electroporation
Image of Cells After Lentiviral Infection
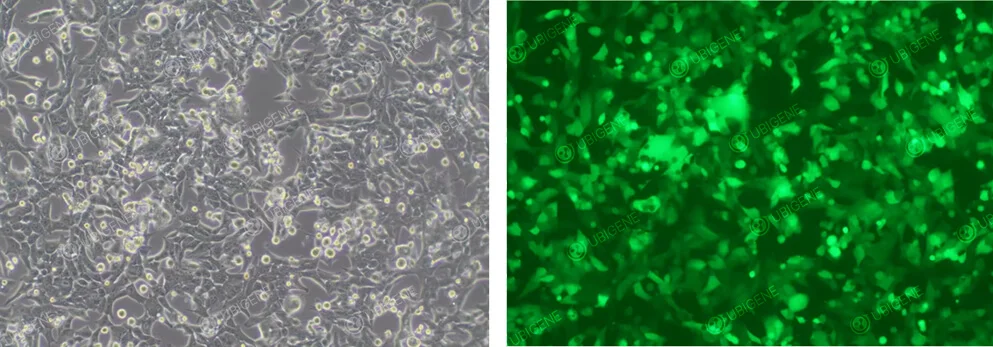
Figure 4. GL261 Cells After Lentiviral Infection








