Essentials | A Comprehensive Overview of hPSCs-Based Models for Neurodegenerative Diseases


Essentials | A Comprehensive Overview of hPSCs-Based Models for Neurodegenerative Diseases

Neurodegenerative diseases (NDDs) are a class of neurological disorders characterized by the progressive and irreversible structural and functional damage or loss of neurons in the central nervous system (CNS) or peripheral nervous system (PNS). Their etiology is complex and involves interactions among multiple factors, including genetics, environment, and aging, ultimately leading to cognitive impairments, motor dysfunctions, sensory abnormalities, and even death. With the rapid aging of the global population, common NDDs such as Alzheimer's disease (AD) and Parkinson's disease (PD) have become increasingly serious public health challenges. In this issue, we will take a deep dive into the panorama of how human pluripotent stem cells (hPSCs) assist in modeling neurodegenerative diseases from cellular mechanisms to disease manifestations. We will focus on modeling strategies, research progress, and prospects for clinical translation. We will also highlight emerging innovations that are shaping the future of neurological disease research.
Research Methods for Neurodegenerative Diseases
Over the past few decades, research on neurodegenerative diseases (NDDs) has identified numerous genetic factors and biochemical processes highly correlated with these conditions. Typical characteristics of NDDs include pathological protein aggregation, synaptic and neuronal network dysfunction, protein homeostasis abnormalities, cytoskeletal changes, energy metabolism disturbances, nucleic acid deficits, inflammation, and neuronal cell death. These core findings are largely attributed to a diverse range of pathological research methods for NDDs. In the early stages of research, common approaches included autopsy brain tissue analysis and pathological studies based on animal models. Through brain tissue analysis during autopsy, we can intuitively observe the pathological features at the terminal stage of human diseases, such as protein deposits and neuronal loss. However, this method cannot flexibly study the dynamic development of the entire disease process, particularly the early pathological changes, and it is challenging to validate the causal mechanisms linking disease occurrence to these pathological features, as it can only relatively limit elucidation of the correlation between the two.
In contrast, methods based on animal models have achieved more impressive breakthroughs in elucidating the pathogenic mechanisms of NDDs in recent years. Commonly used model organisms include mice, rats, fruit flies, and pigs. The use of these model organisms simulates the overall physiological environment of the nervous system and allows for observation of the impacts of complex factors such as the blood-brain barrier, immune system, and neural networks on the onset of NDDs. Additionally, they are suitable for behavioral studies and in vivo drug efficacy evaluations. However, due to significant differences between humans and these model organisms in brain structure, gene expression, and metabolic pathways, many animal models struggle to fully replicate human-specific pathological features, such as neurofibrillary tangles in Alzheimer’s disease (AD) and Lewy bodies in Parkinson’s disease (PD). Furthermore, some experimental results and phenomena derived from animal models are difficult to replicate in human systems.
Over the past two decades, human pluripotent stem cells (hPSCs) have made unprecedented breakthroughs and advances in embryonic development and regenerative medicine. These advances have facilitated their widespread application in neuroscience, enhancing understanding of the pathogenesis of related diseases and promoting exploration of new therapeutic targets and methods. Particularly, with the development of reprogramming technology and continuous optimization of neuron differentiation technology, the application of induced pluripotent stem cells (iPSCs) in research on the mechanisms of neurological diseases and the construction of various models of neurodegenerative diseases has been significantly advanced.
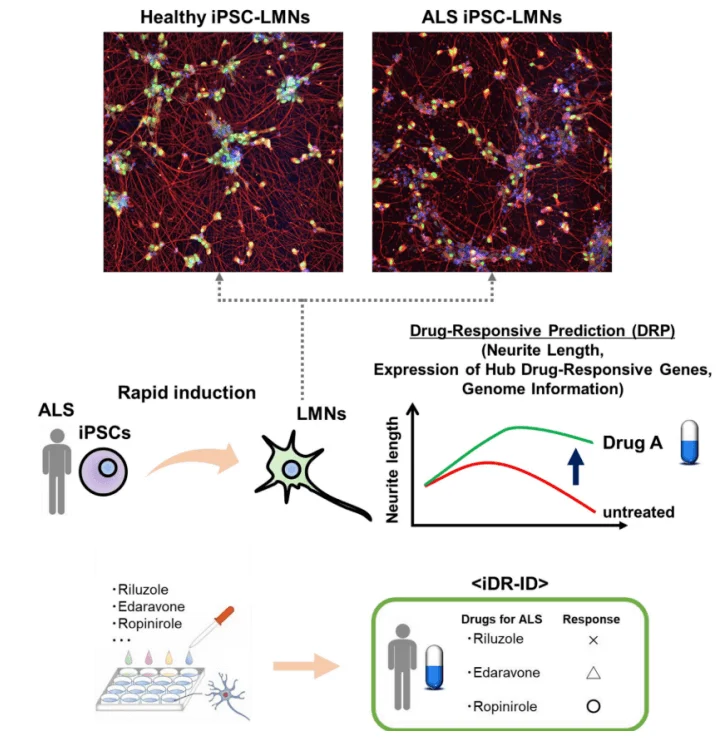
Figure 1. Application of disease modeling and drug efficacy evaluation based on hPSCs [1]
Potential Applications of hPSCs in Neurodegenerative Disease Research
Human pluripotent stem cells (hPSCs) mainly consist of human embryonic stem cells (hESCs) and human induced pluripotent stem cells (hiPSCs). They possess pluripotency, self-renewal capabilities, and the ability to differentiate into multiple cell types. Therefore, by utilizing hPSCs that can proliferate for extended periods, it is theoretically possible to obtain a large number of neural lineage cells or related brain organoids for disease modeling. These can be used for studying corresponding pathological mechanisms or assessing drug efficacy. Among them, hiPSCs can be derived from patients' somatic cells, maintaining the same genetic background as the patients. Through specific differentiation techniques, hiPSCs can be induced into disease cell models or organoid models that retain the patient's genetic characteristics. On the other hand, hESCs or hiPSCs from healthy donors can also be genetically edited to knockout specific genes or introduce point mutations, further constructing disease models by simulating pathological genetic features.
So, what types of neural lineage cells can be obtained through induced differentiation of hPSCs? For different neurodegenerative diseases, which types of neural cells should be selected for application, and what are the specific pathological phenotypes of these cells in the context of the corresponding degenerative diseases? Today, Ubigene will address these questions one by one, providing useful insights for researchers in the fields of neuroscience and neurodegenerative disease research.
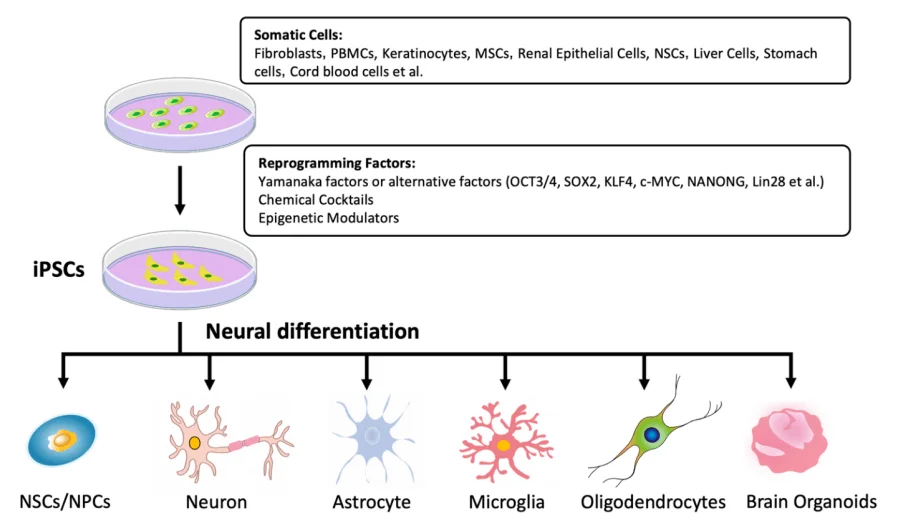
Figure 2. The neural lineage differentiation potential of iPSCs
Neurogenic Potential of hPSCs
In numerous neurodegenerative diseases, the abnormal phenotypes of neurons, glial cells, and other neural lineage cells are closely related to the onset of the diseases. However, obtaining these cells has always posed tremendous challenges. The advent of hPSCs (human pluripotent stem cells) has significantly alleviated this problem. Due to their multi-directional differentiation potential and the continuous optimization of neural directed differentiation methods, various types of neural cells and organoids derived from hPSCs have been widely applied in the field of neuroscience. They particularly show great potential in establishing in vitro disease models, providing an ideal system and platform for studying the pathogenesis of neurodegenerative diseases and drug screening. Below, we have listed the main differentiation types and common differentiation methods for neural lineage cells and brain organoids based on hPSCs.
| Cell Type/Organ Type | Differentiation Method |
|---|---|
| NSCs/NPCs |
(1) Embryoid body formation (2) Co-culturing with neural induction feeder layer (3) Double SMAD signaling inhibition |
| Neurons |
(1) Adding neurotrophic factors at NSC stage (2) Direct transdifferentiation |
| Astrocytes |
(1) Adding inducing factors at NPC stage (2) Adding inducing factors at oligodendrocyte precursor cell stage |
| Microglia |
(1) Induction from mesodermal progenitor cells (2) Overexpression of transcription factors |
| Oligodendrocytes |
(1) Stepwise induction (2) Overexpression of transcription factors |
| Brain organoids (cortex, hippocampus, etc.) |
(1) Spontaneous differentiation induction (2) Directed differentiation induction |
Table 1. Neural differentiation potential of hPSCs
Modeling Neurodegenerative Diseases Based on hPSCs
As mentioned earlier, iPSCs and their derivatives are genomically identical to the donor, providing a unique in vitro research tool for neurodegenerative diseases that lack suitable models. Additionally, for sporadic cases, gene editing can be performed on hESCs or healthy individuals' hiPSCs to knock out genes or introduce pathogenic mutations, simulating pathological genetic features. The establishment of such models is crucial for elucidating disease mechanisms and developing targeted therapies. Below, I will illustrate the commonly used cellular or organoid disease models for three representative neurodegenerative diseases—Alzheimer's disease (AD), Parkinson's disease, and Huntington's disease (HD)—along with their corresponding pathological features.
Alzheimer's Disease (AD)
Statistically, up to 95% of AD patients have sporadic cases, while familial cases account for only 5%, with the latter being associated with mutations in genes such as APP, PSEN1, and PSEN2. The main risk factor for sporadic AD is the APOE4 allele. The core pathological hallmarks of AD are the accumulation of β-amyloid (Aβ) in neurons and the hyperphosphorylation of Tau protein leading to neurofibrillary tangles; however, the causal relationship between these pathological features and the onset of AD remains unclear. Moreover, the neurons derived from hPSCs not only replicate these two core characteristics but also exhibit other pathological phenotypes, such as activation of GSK3β, abnormal electrophysiological activity in neurons, enhanced oxidative stress, increased reactive oxygen species production, endoplasmic reticulum dysfunction, and mitochondrial abnormalities. In addition to neurons, researchers have also utilized other hPSC-derived in vitro models to study AD, including neural stem/progenitor cells (NSCs/NPCs), microglia, astrocytes, oligodendrocytes, and brain organoids. Below, we will summarize the pathological features of various in vitro research models used in AD studies:
| Model Type | AD Model Pathological Features |
|---|---|
| Neurons |
(1) Aβ accumulation (2) Tau hyperphosphorylation (3) GSK-3β signaling activation (4) Potential abnormalities (5) Increased oxidative stress and reactive oxygen species production (6) Endoplasmic reticulum dysfunction (7) Mitochondrial abnormalities (8) Increased sensitivity to Aβ42 |
| NSCs/NPCs (Neural Stem Cells/Neural Progenitor Cells) |
(1) Low APP and Aβ expression levels (2) Significantly impaired proliferation and self-renewal capabilities (3) Increased expression of neurodevelopmental-related genes |
| Astrocytes |
(1) Abnormal morphology (2) Increased release of Aβ42 (3) Impaired clearance ability (4) Dysregulated cytokine secretion (5) Calcium homeostasis imbalance (6) Elevated levels of reactive oxygen species |
| Microglia |
(1) Reduced phagocytic ability towards Aβ and Tau oligomers (2) Enhanced neuroinflammation |
| Oligodendrocytes |
(1) Morphological defects (2) Impaired proliferation and function (3) Reduced neuronal support capacity (4) Defects in myelination |
| Brain Organoids |
(1) Aβ deposition (2) Formation of neurofibrillary tangles (3) Enhanced Tau phosphorylation (4) Abnormalities in the endoplasmic reticulum |
Table 2. In Vitro Studies of Alzheimer's Disease Neuronal Models and Their Pathological Features
Parkinson's Disease (PD)
PD is the second most common neurodegenerative disease, after Alzheimer's disease. Approximately 10% of PD patients are diagnosed with familial Parkinson's disease, while the remaining cases are classified as sporadic Parkinson's disease. The main pathological feature of PD is the progressive loss of dopaminergic (DA) neurons, accompanied by the accumulation of alpha-synuclein and the formation of Lewy bodies. Genetic mutations such as SNCA, PARK2, PINK1, and LRRK2 are closely associated with the pathogenesis of PD, although their exact roles are still to be explored. It is evident that DA neurons serve as the most effective and commonly used in vitro research models for studying the pathological mechanisms of PD and screening potential therapeutic drugs. Furthermore, other neural lineage cells, such as astrocytes, microglia, and neural stem cells, as well as 3D brain organoid models, have also received widespread attention and application in the study of PD pathogenesis. Similarly, the following list provides commonly used in vitro models for studying this degenerative disease and their corresponding pathological phenotypes:
| Model Type | PD Model Pathological Features |
|---|---|
| DA Neurons |
(1) Accumulation of α-synuclein (2) Mitochondrial dysfunction (3) Increased sensitivity to oxidative stress/endoplasmic reticulum stress (4) Synaptic loss (5) Neuronal death (6) Increased apoptosis (7) Impaired neuronal homeostasis (8) Defects in mitochondrial autophagy (9) Elevated levels of reactive oxygen species (ROS) (10) Epigenetic changes |
| Neural Stem Cells/Neural Progenitor Cells (NSCs/NPCs) |
(1) Reduced differentiation efficiency (2) Developmental defects |
| Astrocytes |
(1) Accumulation of α-synuclein (2) Autophagy dysregulation (3) Abnormal mitochondrial morphology/activity (4) Reduced cell viability (5) Oxidative stress-induced degeneration of DA neurons |
| Microglia |
(1) Accumulation of α-synuclein (2) Impaired phagocytic ability (3) Enhanced neuroinflammation (4) Disruption of the morphology/function of dopaminergic neurons |
| Brain Organoids |
(1) Increased death of dopaminergic neurons (2) Reduced differentiation potential (3) Decreased dendritic complexity (4) Neurodevelopmental defects |
Table 3. In Vitro Studies of Neuronal Models for PD and Their Pathological Features
Huntington's disease (HD)
HD is an autosomal dominant neurodegenerative disorder characterized by progressive motor dysfunction, cognitive decline, and psychiatric disturbances. HD is caused by an abnormal expansion of the CAG trinucleotide repeat sequence within the first exon of the huntingtin gene (HTT) located on chromosome 4. Statistics indicate that the length of the CAG repeat sequence is negatively correlated with the age of disease onset and positively correlated with the severity of the disease. Pathologically, hallmark features of HD include: aggregation of the mutant HTT protein, progressive degeneration of the striatum with selective loss of medium spiny neurons (MSNs), disruption of neurotransmitter homeostasis, mitochondrial dysfunction, and significant extrapyramidal motor abnormalities such as chorea and dystonia. The loss of GABAergic MSNs in the striatum represents the most characteristic and prominent phenotype of HD. These phenotypic feature, along with other phenotypes observed in in vitro research models, is summarized as follows:
| Model Type | PD Model Pathological Features |
|---|---|
| NSCs/NPCs |
(1) Expression of mutant HTT protein with CAG repeat expansion (2) Transcriptional dysregulation (3) Abnormal electrophysiological characteristics (4) Increased apoptosis (5) Enhanced sensitivity to neurotrophic factor deprivation (6) Increased apoptosis (7) Impaired neuronal homeostasis (8) Defects in mitophagy (9) Elevated levels of reactive oxygen species (ROS) (10) Epigenetic changes |
| Forebrain Neurons |
(1) Aggregation of mutant HTT protein (2) Synaptic dysfunction (3) Transcriptional dysregulation |
| MSNs |
(1) Aggregation of mutant HTT protein (2) Increased lysosomal and autophagosomal activity (3) Nuclear invagination (4) Caspase activation (5) Accelerated neuronal death during aging (6) Mitochondrial dysfunction |
| Astrocytes |
(1) Vacuolar phenotype (2) Enhanced sensitivity to neurotrophic factor deprivation (3) Impaired neuroprotective function |
| Glial Cells (General) | Enhanced sensitivity to neurotrophic factor deprivation |
Table 4. In Vitro Neurocellular Models of Huntington's Disease and Their Pathological Features
After reviewing the modeling strategies of the three major neurodegenerative diseases (NDDs), has everyone gained a more systematic understanding of research on neurological diseases? Currently, numerous studies have demonstrated that neurons or neural stem cells derived from human pluripotent stem cells (hPSCs) can effectively model the specific pathological features of NDD patients when combined with gene editing to introduce potential pathogenic mutations or repeat sequences. Alternatively, neural lineage cells induced from iPSCs derived from NDD patients can also serve as effective models. It can even be said that many of the disease mechanisms and treatment strategies revealed today have arisen from in vitro research models derived from hPSCs. In fact, besides the three major NDDs, neural lineage cells such as neurons and glial cells based on hPSCs have also shown remarkable potential in the mechanism research of spinocerebellar ataxia (SCA) and spinal muscular atrophy (SMA), exhibiting disease-specific phenotypes and susceptibility, making them effective and reliable in vitro research models.
However, although the differentiation of hPSCs into neurons or neural stem cells is an important approach for studying the development of the nervous system, disease modeling, drug screening, and targeted therapy, this process still faces numerous difficulties and challenges, such as:
1.Differentiation Efficiency and Heterogeneity
The neural differentiation process often results in a large mixture of cell populations, including target neurons, neural stem cells, and various non-target neuronal subtypes, as well as incompletely differentiated cells and even residual pluripotent stem cells. Achieving a high-purity, specific type of cell population is challenging. Moreover, the differentiation propensity and efficiency may vary significantly between different hPSC lines and even between different batches from the same line, making batch-to-batch stability difficult to control.
2.Functional Maturity
Without stable and mature in vitro differentiation protocols, the resulting neurons may exhibit immature characteristics in protein expression, synaptic connections, electrophysiological properties, metabolic levels, and gene expression profiles, and may not be suitable as effective in vitro disease models.
3.Standardization and Reproducibility of Procedures
Differentiation protocols usually involve multiple stages, various growth factors, small molecules, and matrices, making the steps complicated and requiring high technical proficiency. Additionally, determining how to define the differentiation state at each stage, as well as the criteria for complete differentiation and functional maturity, needs to be managed by technicians with relevant expertise and extensive experience throughout the process.
4.Cost of Induction
The costs associated with high-quality hPSC culture and neural differentiation, which require special culture media, growth factors, small molecules, and matrix gels, tend to be quite high, thereby limiting experimental flexibility and protocol design.
Currently, Ubigene Biosciences has launched technical services and related products for the differentiating of hPSCs (including hESCs and hiPSCs) into neural stem cells,neural progenitor cells, and neurons (cortical and forebrain). The related differentiation protocols and effectiveness illustrations are as follows:
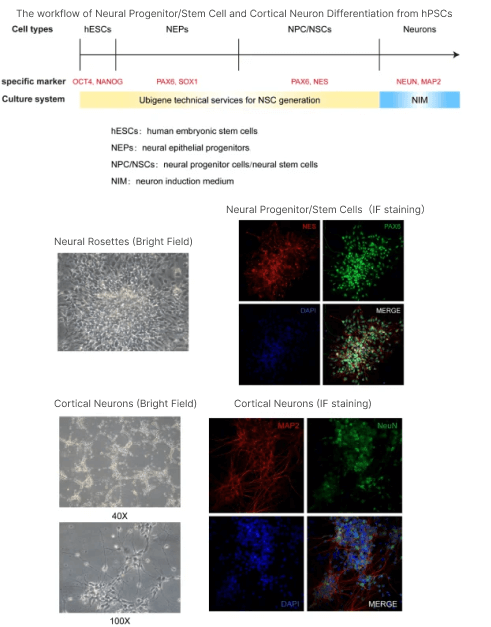
Figure 3. Experimental process and detection results of Ubigene Biosciences hPSC neural differentiation technology services.
Currently, the differentiation of human pluripotent stem cells (hPSCs) into neural system cells still faces many challenges, including low differentiation efficiency, process instability, and poor reproducibility. In response to these technical bottlenecks, Yuanjing Biotechnology relies on its independently developed neural differentiation technology system, combined with a mature and stable stem cell culture platform, to provide researchers with a one-stop solution for efficiently inducing differentiation of hPSCs into neural stem cells (NSCs) or neurons.
Additionally, Ubigene possesses an advanced CRISPR gene editing platform enabling precise modification of various targets, providing strong technical support for research projects such as neural system development studies, disease model construction, drug screening, and pharmacological evaluation.
1. Efficient Neural Differentiation Program and Precise Stage Control
Significantly improves the purity and yield of differentiation into neural stem cells and target neurons, effectively reducing mixed cell populations. A strict standardized culture and induction system, combined with advanced real-time detection and quality control methods, greatly enhances the stability and consistency of differentiation outcomes across batches.
2.Functional Assurance of Neural Stem Cells/Neurons
Equipped with comprehensive detection methods for neural lineage cells, the system ensures the proper morphology and specific marker expression of the obtained neural stem cells and neurons, as well as the functional maturation of neurons, thereby laying a solid foundation for their research applications.
3. Seamless Integration of Gene Editing
Equipped with a self-developed and stable gene editing platform, efficiently realizing gene knockout, point mutation, and knock-in editing projects, easily facilitating gene function studies and disease modeling, enhancing the efficiency and success rate of projects.
4.Professional Technical Team Suppor
Boasting a PhD-level professional technical team, the company ensures thorough oversight throughout the entire project process, with project personnel possessing extensive experience in hPSC culture and stem cell differentiation.
This concludes the technical topic sharing for this session! If you are interested in hPSC neural differentiation or CRISPR gene editing, or if you have specific technical needs in related research, feel free to leave your contact information or contact Ubigene for inquiries. We will provide you with professional support and customized solutions as soon as possible, to help you advance your research!
References
[1] Okano H, Morimoto S. iPSC-based disease modeling and drug discovery in cardinal neurodegenerative disorders. Cell Stem Cell. 2022 Feb 3;29(2):189-208.
[2] Guo X, Wang X, Wang J, Ma M, Ren Q. Current Development of iPSC-Based Modeling in Neurodegenerative Diseases. Int J Mol Sci. 2025 Apr 16;26(8):3774.








