Uncovering Dual Drug Resistance in Pancreatic Cancer: CRISPR Screens Identify JUN as a Common Mediator of Resistance to SHP2/ERK and RAS (ON) Inhibitors


Introduction
Pancreatic cancer is among the most lethal malignancies worldwide, with KRAS mutations present in over 85% of pancreatic ductal adenocarcinomas (PDAC), making it a central oncogenic driver.Although combined MAPK pathway-targeted therapies (e.g., SHP2 plus ERK inhibitors) have entered clinical evaluation, acquired or intrinsic resistance remains a major barrier to efficacy. The team led by Rene Bernards and Sara Mainardi at the Netherlands Cancer Institute employed genome-wide CRISPR knockout screens followed by functional validation to uncover the mechanistic basis of MAPK inhibitor resistance in KRAS-mutant PDAC

Research Background
Pancreatic cancer has a 5-year survival rate of only ~10%, primarily due to late-stage diagnosis and limited therapeutic options. Conventional chemotherapy offers modest benefit, and targeted therapies are effective only in a small subset of patients with homologous recombination defects.KRAS is the dominant oncogenic driver in pancreatic ductal adenocarcinoma (PDAC).
Historically considered “undruggable” due to structural constraints, recent advances have led to the development and clinical approval of KRAS^G12C-specific inhibitors for non-small cell lung cancer. However, inhibitors targeting KRAS^G12D, the most prevalent mutation in PDAC, remain in early clinical stages.Currently, the mechanisms of resistance to combination therapies targeting the MAPK pathway, such as SHP2+ERK or RAS(ON) multi-selective inhibitors, are largely unknown. A systematic identification of resistance mediators and underlying pathways is urgently needed to guide clinical strategies for overcoming therapy resistance in KRAS-mutant PDAC.
Research Objective
The study aimed to systematically identify mediators of resistance to SHP2+ERK and SHP2+RAS(ON) inhibitor combinations in KRAS-mutant PDAC, elucidate their molecular mechanisms, and validate whether targeting these pathways could reverse resistance. The ultimate goal was to provide novel therapeutic targets and strategies for precision treatment of KRAS-driven pancreatic cancer.
Research Methods
-
Cell Line Models:
- KRAS-mutant PDAC cell lines (YAPC-1, ASPC-1, Panc 10.05, Panc-1, MiaPaCa-2) were used.MiaPaCa-2 cells were continuously exposed to SHP2+ERK inhibitors (RMC-4550 + LY3214996) or SHP2+RAS(ON) inhibitors (RMC-4550 + RMC-6236) to generate spontaneous resistant lines (Resistant, Resistant_1).
- PTEN knockout single clones were generated via lentiviral sgRNA transduction, and JUN-overexpressing lines were constructed via lentiviral vectors.All cell lines were validated by STR profiling and mycoplasma testing; HEK293T cells were used for lentivirus packaging.
- Genome-wide CRISPR Knockout Screening: Panc 10.05 cells were transduced with the Brunello genome-wide sgRNA library.Post puromycin selection, cells were treated with low (2 μM RMC-4550 + 2 μM LY3214996) and high (4 μM RMC-4550 + 4 μM LY3214996) doses, alongside untreated controls.sgRNAs were recovered and sequenced; DESeq2 and MAGeCK RRA analyses identified candidate resistance genes.
- Focused CRISPR Screening: A custom sgRNA library targeting 167 candidate genes from the primary screen was applied in 4 KRAS-mutant PDAC lines at 800× coverage.STRING protein interaction analysis was used to identify core resistance candidates.
- Molecular Biology Experiments: Western blot (WB) and immunohistochemistry (IHC) were used to assess pathway activation.
- Animal Experiments: Panc 10.05 PTEN-knockout cells (5×10⁶) were implanted subcutaneously into NSG mice.Tumors were treated with: vehicle, HRX-0233, SHP2+ERK inhibitors, or triple therapy (HRX-0233 + SHP2+ERK inhibitors) by oral gavage.Tumor volumes were monitored to validate the in vivo efficacy of targeted interventions.
Research Workflow
- Identification of resistance mediators: Two-step CRISPR screening (genome-wide + focused) in KRAS-mutant PDAC identified PTEN, DET1, COP1, among others, as resistance-associated genes.STRING analysis highlighted PTEN as a central hub.
- Validation of PTEN-mediated resistance: PTEN knockout cells showed activation of the PI3K-AKT-mTOR pathway and resistance to SHP2+ERK inhibitors.Spontaneous resistant lines displayed similar pathway activation, confirming mechanistic relevance.
- Linking mTOR activation to JUN expression: PTEN-knockout and spontaneous resistant cells exhibited high JUN expression, indicating mTOR drives JUN upregulation.JUN acts as a key downstream effector of mTOR.
- Confirming JUN as a master resistance mediator: JUN overexpression independently conferred resistance. Targeting JNK/MAP2K4 to downregulate JUN restored SHP2+ERK inhibitor sensitivity.
- In vivo validation of resistance reversal: In PTEN-knockout xenografts, combining HRX-0233 (MAP2K4 inhibitor) with SHP2+ERK inhibitors effectively suppressed tumor growth, reversing resistance.
- Expanding JUN's role in resistance: JUN also mediated resistance to SHP2+RAS(ON) multi-selective inhibitors, confirming it as a broad-spectrum resistance node.
Key Results
1. CRISPR screens reveal mediators of SHP2+ERK inhibitor resistance in KRAS-mutant PDAC
The research team first performed genome-wide CRISPR knockout screens in Panc 10.05 cells, applying SHP2+ERK inhibitor combinations at varying concentrations as selective pressure. This primary screen identified multiple resistance-associated genes.
- Shared core genes between low- and high-dose treatment groups included PTEN, PPP6C, PPP2R4, among a total of six candidates.
- The high-dose group, under more stringent selection, yielded fewer hits, reflecting stronger selective pressure.
To refine the findings, the team conducted focused CRISPR screens across four KRAS-mutant PDAC cell lines, revealing cell line-specific resistance profiles:
- MiaPaCa-2 and ASPC-1: DET1 and COP1 were consistently enriched. These two proteins form an E3 ubiquitin ligase complex, which regulates the stability of JUN, a key downstream effector of resistance.
- Panc 10.05 and Panc-1: Shared resistance genes included PTEN and CYB5R4.
- PPP2R4, although enriched in three cell lines, did not validate as a functional mediator when tested individually with sgRNAs.
Subsequent STRING protein interaction analysis revealed PTEN as the central hub connecting the identified resistance genes. This highlighted PTEN as the primary candidate for in-depth mechanistic studies, positioning it at the core of SHP2+ERK inhibitor resistance in KRAS-mutant PDAC.
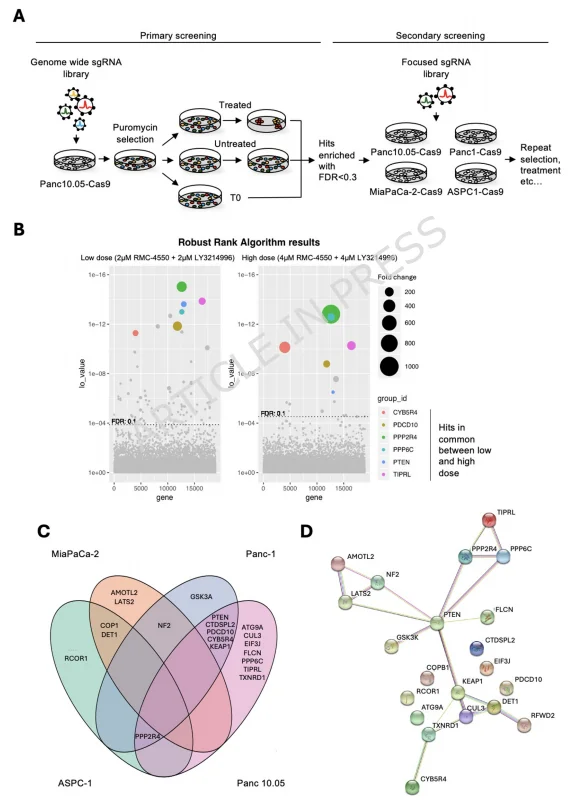
Figure 1. CRISPR Gene Screens Identify Mediators of SHP2+ERK Inhibitor Resistance in KRAS-Mutant PDAC
2. PTEN Loss or Spontaneous PI3K-AKT-mTOR Activation Mediates Resistance to SHP2+ERK Inhibitors
The research team next investigated the role of PTEN loss and PI3K-AKT-mTOR pathway activation in mediating resistance to SHP2+ERK inhibitors. Key findings in PTEN knockout cells (Panc 10.05, Panc-1, ASPC-1):
- Pathway activation: Increased levels of p-AKT and p-S6RP confirmed PI3K-AKT-mTOR hyperactivation.
- Drug resistance: PTEN knockout significantly enhanced resistance to SHP2+ERK inhibitors, with suppressed apoptosis upon drug treatment.
Spontaneous resistance model (MiaPaCa-2):
- Resistant clones did not show MAPK reactivation (low p-RSK), but exhibited reduced PTEN expression and elevated p-S6RP, similar to PTEN knockout cells.
- These cells demonstrated a “drug addiction” phenotype, with proliferation inhibited after drug withdrawal.
- Single-cell clones of the resistant population retained both resistance and PI3K-AKT-mTOR activation.
In vivo validation: MiaPaCa-2 xenografts treated with SHP2+ERK inhibitors showed enhanced p-S6RP staining, indicating mTOR activation contributes to acquired resistance during therapy.
Therapeutic interventions:
- mTOR inhibitor AZD8055 suppressed proliferation of resistant cells.
- Combination of AZD8055 + SHP2+ERK inhibitors effectively inhibited growth of parental and resistant cells.
- Partial resistance remained in some lines (MiaPaCa-2, Panc-1), suggesting mTOR-independent resistance mechanisms exist.
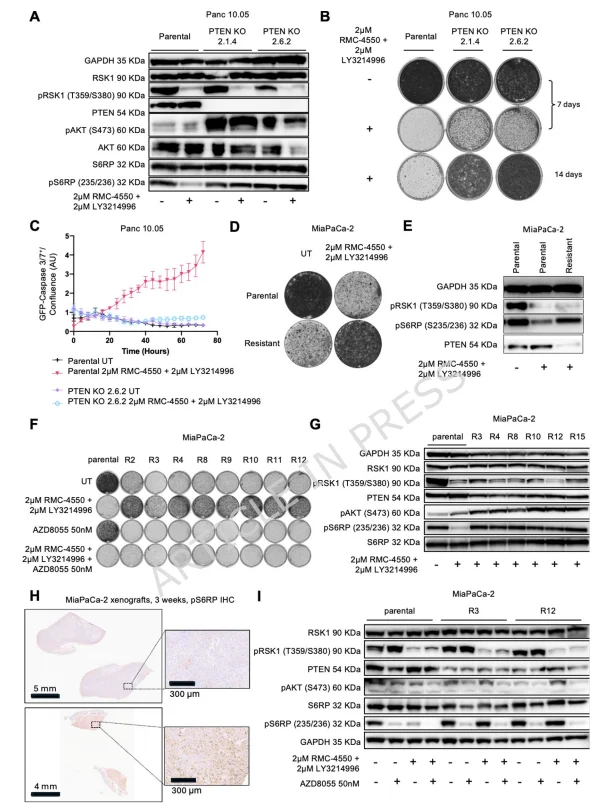
Figure 2. PTEN Loss or PI3K-AKT-mTOR Pathway Activation Drives Resistance to SHP2+ERK Inhibition
3. JUN as the Downstream Master Mediator of MAPK Inhibitor Resistance in KRAS-Mutant PDAC
The team further discovered that in MiaPaCa-2 spontaneous resistant clones and PTEN knockout cells, both total JUN and phosphorylated JUN (p-JUN) levels were markedly elevated and remained unaffected by SHP2+ERK inhibitor treatment, whereas in parental cells, p-JUN levels decreased in response to the drug.RNA-seq analysis revealed dysregulation of the AP-1 transcription factor network in PTEN knockout cells, with significant upregulation of JUN and downregulation of the inhibitory AP-1 family member BATF, indicating a shift of AP-1 activity toward pro-proliferative signaling.
Pharmacological interventions showed that the mTOR inhibitor AZD8055 simultaneously suppressed S6RP phosphorylation and both total and phosphorylated JUN levels. Combining AZD8055 with SHP2+ERK inhibitors restored drug sensitivity in PTEN knockout cells. Furthermore, targeting upstream JUN regulators—JNK inhibitor SP600125 or MAP2K4 inhibitor HRX-0233—reduced p-JUN levels and re-sensitized resistant cells to SHP2+ERK inhibition. Conversely, JUN overexpression alone was sufficient to confer resistance, independent of MAPK pathway reactivation.This work establishes JUN as a central downstream mediator of MAPK inhibitor resistance in KRAS-mutant pancreatic cancer, highlighting both its mechanistic role and therapeutic potential as a target for overcoming resistance.
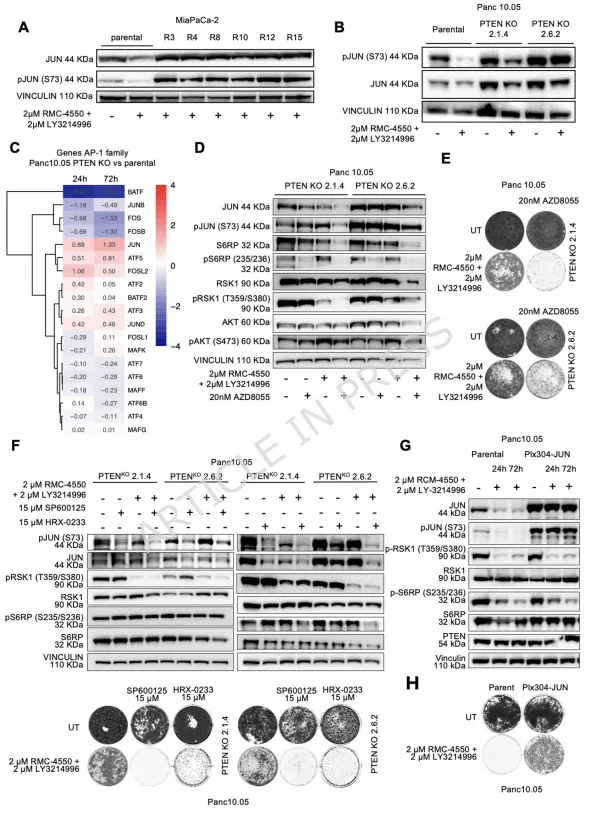
Figure 3. JUN Functions as the Downstream Master Mediator of MAPK Inhibitor Resistance in KRAS-Mutant PDAC
4. JUN as an In Vivo Therapeutic Target and Mediator of Resistance to SHP2+RAS(ON) Inhibitors
In vivo studies demonstrated that Panc 10.05 PTEN knockout xenografts were resistant to SHP2+ERK inhibitors. However, co-treatment with the MAP2K4 inhibitor HRX-0233 effectively suppressed tumor growth, confirming that targeting JUN can reverse in vivo resistance.In vitro, the combination of SHP2 inhibitor RMC-4550 and RAS(ON) multi-selective inhibitor RMC-6236 exhibited strong synergistic anti-tumor activity in PDAC cell lines. To investigate resistance mechanisms, the team generated MiaPaCa-2 spontaneous resistant cells (Resistant_1). Key observations:
- Resistant_1 cells displayed a milder resistance phenotype compared to SHP2+ERK-resistant cells and remained proliferative after drug withdrawal.
- PTEN expression was downregulated, but the mTOR pathway was not activated, while p-JUN levels were markedly elevated, indicating that JUN mediates resistance in an mTOR-independent manner.
- JUN overexpression alone conferred resistance to SHP2+RAS(ON) inhibitors in Panc 10.05 and Panc-1 cells, independent of MAPK pathway reactivation.
These results establish JUN as a broad-spectrum core node of drug resistance in KRAS-mutant PDAC and validate it as a therapeutically actionable in vivo target.
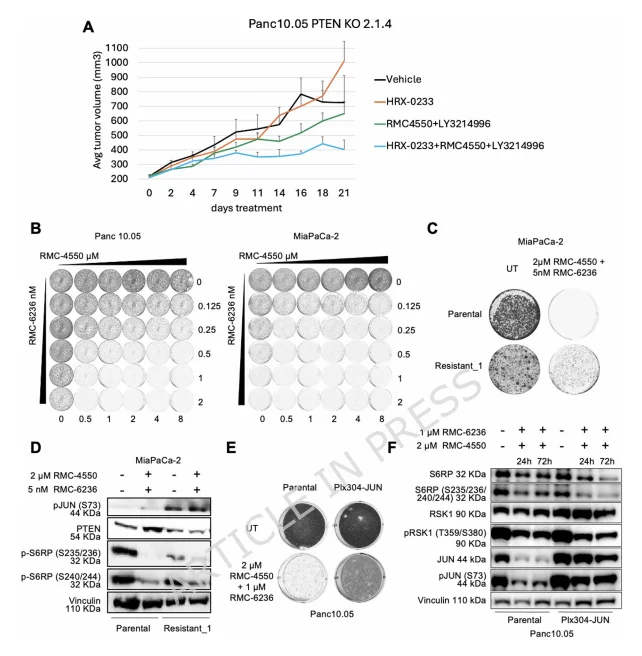
Figure 4. JUN Functions as an In Vivo Resistance Mediator and Confers Resistance to SHP2+RAS(ON) Inhibitors
Summary
This study systematically elucidated the mechanisms of resistance to MAPK inhibition in KRAS-mutant pancreatic ductal adenocarcinoma (PDAC) through genome-wide CRISPR screening combined with in vitro and in vivo functional validation. Key findings include:
- PTEN loss–mediated activation of the PI3K-AKT-mTOR pathway and elevated JUN expression were identified as the core resistance mechanisms.
- JUN was confirmed as the most downstream and critical mediator of MAPK inhibitor resistance.
- Targeting MAP2K4 to inhibit JUN effectively reversed resistance in vivo, providing a theoretical basis and actionable target for optimizing combination therapies in KRAS-mutant PDAC.
These findings not only deepen our understanding of molecular mechanisms underlying targeted therapy resistance in pancreatic cancer but also lay the foundation for developing more effective combination treatment strategies to improve patient outcomes.
Ubigene CRISPR-iScreen™ Library
CRISPR-iScreen™ is an innovative platform independently developed by Ubigene, designed to enable high-efficiency CRISPR-based functional screening.
- Comprehensive coverage: Ubigene offers over 40 ready-to-use CRISPR libraries covering human, mouse, green monkey, and pig, including genome-wide knockout, CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa) libraries.
- Focused sublibraries: Specialized libraries targeting kinases, cell cycle regulators, membrane proteins, metabolic genes, and other functional gene sets facilitate streamlined target discovery.
- Ready-to-use resources: More than 150 CRISPR library lentivirus and 400+ CRISPR library cell pools are available in stock. Ubigene also provides end-to-end CRISPR screening services (both in vitro and in vivo), supporting your research needs at every stage.
iScreenAnlys™ CRISPR Library Analysis platform is available for all users, offering flexible selection among Drug-Z, MAGeCK-MLE, and MAGeCK-RRA algorithms for one-click analysis of screening data, helping accelerate your research workflow.
Contact us for more information >>>Reference
Mulero-Sánchez A, Bosma A, Visuvasam B, Pouliopoulou N, van de Ven M, Proost N, Boeije M, Lieftink C, Beijersbergen R, Bernards R, Mainardi S. CRISPR knockout screens reveal JUN as the master mediator of resistance to MAPK inhibition in KRAS-mutant pancreatic cancer. J Exp Clin Cancer Res. 2026 Jan 22. doi: 10.1186/s13046-025-03616-z. Epub ahead of print. PMID: 41572361.
 Promotions
Promotions 








