CRISPR Screening Guide: Libraries, Methods & Workflows


Introduction to CRISPR Screening
CRISPR-based screening is a robust and versatile high-throughput method for functional genomics research. Compared with traditional RNAi techniques, CRISPR screens provide higher specificity, lower off-target effects, and more efficient gene knockout, enabling accurate and reproducible insights into gene function.
Scope of This Guide
In this article, Ubigene offers a concise overview of CRISPR screening, covering its fundamental concepts, classification types, guide RNA library selection strategies, screening workflows, and practical implementation. This guide aims to inspire new research ideas while providing actionable insights for designing and executing CRISPR-based screens.
What is CRISPR Screening?
CRISPR screening is a high-throughput gene screening method based on CRISPR/Cas9 technology. In this approach, sgRNAs are synthesized and packaged into lentiviruses, which are then used to infect cells at a low multiplicity of infection (MOI < 0.3) to ensure that each cell receives only one sgRNA. Cells are subsequently subjected to a specific treatment or condition—such as drug exposure, hypoxia, viral infection, or assessment of cell viability. After selection, the cells are collected for next-generation sequencing (NGS) to analyze the enrichment or depletion of sgRNAs between experimental and control groups, identifying genes related to the screened phenotype.
Compared with cDNA- or RNAi-based screens, CRISPR screen offers higher versatility, lower background noise, more efficient gene knockout, and fewer off-target effects, making it the preferred method for large-scale functional genomic studies.
Advantages and Flexibility
These screens are highly scalable and adaptable, supporting genome-wide analysis of:
- Gene essentiality
- Drug resistance mechanisms
- Cellular signaling pathways
Importantly, both loss-of-function and gain-of-function strategies can be applied, providing a comprehensive understanding of gene regulatory networks. CRISPR screens can also be tailored to specific phenotypic readouts, such as cell viability, apoptosis, immune response, or drug sensitivity, making them indispensable for basic research and therapeutic target discovery.
RNAi vs. CRISPR Screening: Two Powerful Tools for Studying Genes
RNA interference (RNAi) and CRISPR screening are two powerful tools that allow scientists to explore how genes control the behavior of cells.
RNAi works by using small RNA molecules (siRNA or shRNA) that attach to a target gene's messenger RNA and trigger its degradation. This reduces the production of the corresponding protein, creating a gene knockdown effect — the gene is still there, but its activity is turned down. Because the suppression is partial and temporary, RNAi is useful for studying essential genes that would kill the cell if completely removed. However, it can sometimes silence unintended genes, leading to off-target effects and variable results.
1. How RNAi Screening Works?
RNA interference (RNAi) is a natural process used by cells to regulate gene expression.It relies on small pieces of RNA, known as small interfering RNA (siRNA) or short hairpin RNA (shRNA), which bind to a gene's messenger RNA (mRNA). Once bound, the mRNA is cut and destroyed, preventing the cell from producing the corresponding protein.
In laboratory RNAi screening, scientists introduce libraries of siRNAs or shRNAs into cells, each designed to target a different gene.By measuring changes in cell behavior—such as growth rate, survival, or resistance to drugs—they can determine which genes are essential for specific cellular processes.
Because RNAi acts at the mRNA level, the effect is temporary and partial.It reduces, but does not completely eliminate, gene activity.
2. How CRISPR Screening Works?
CRISPR-Cas9, on the other hand, is a tool for gene editing at the DNA level.The system uses an enzyme called Cas9 and a short RNA guide (sgRNA) that directs Cas9 to a precise spot in the genome.Once there, Cas9 makes a cut in the DNA.When the cell tries to repair the break, small mutations are introduced that disrupt the gene permanently.
In CRISPR screening, thousands of sgRNAs are used in parallel, allowing researchers to knock out nearly every gene in the genome and identify which ones are important for a given cellular function.Because CRISPR editing causes permanent genetic changes, the resulting data tend to be more reliable and reproducible than those from RNAi.
3. Side-by-Side Comparison
| Feature | RNAi Screening | CRISPR Screening |
|---|---|---|
| Mechanism | Degrades mRNA using siRNA/shRNA | Cuts DNA using Cas9 and sgRNA |
| Level of Action | mRNA level | DNA level |
| Effect on Gene | Knockdown (partial silencing) | Knockout (complete loss) |
| Duration | Temporary and reversible | Permanent |
| Precision | More off-target effects | Generally more specific |
| Efficiency | Often incomplete | High and consistent |
| Reversibility | ✓ Yes | ✗ No |
| Typical Use | Short-term studies, pathway analysis | Genome-wide knockout, essential gene discovery |
4. Applications in Research
Both RNAi and CRISPR screens are fundamental tools in functional genomics, connecting genes to their biological roles.
- RNAi screening was the first technology to make genome-wide loss-of-function studies possible. It has been used to identify genes involved in cancer, viral infection, and drug resistance.
- CRISPR screening has quickly become the new standard. It provides cleaner results, can target non-coding regions of DNA, and is especially useful for finding essential genes or studying genetic interactions that lead to disease. The precision and permanence of CRISPR screens make them powerful engines for biological discovery and therapeutic development, while RNAi remains a useful tool when full gene knockout would be too disruptive.
5. Strengths and Limitations
| Aspect | RNAi | CRISPR |
| Advantages | Simple, reversible, works in many cell types | High precision, stable results, genome-wide reach |
| Limitations | Incomplete silencing, off-target effects | DNA damage risk, irreversible changes |
| Best for | Temporary knockdowns, regulatory studies | Permanent knockouts, large-scale functional screens |
6. Conclusion
RNAi and CRISPR screening have both revolutionized our understanding of how genes shape life. RNAi screening offers a way to gently reduce gene activity—like dimming the lights—while CRISPR screens can switch those lights off completely. Today, CRISPR screening is often the tool of choice for large-scale genetic screens due to its accuracy and durability, yet RNAi remains important for temporary studies or cases where permanent DNA changes are not desirable. Together, these technologies continue to drive breakthroughs in medicine, biotechnology, and beyond.
Learn More: CRISPR vs RNAiTypes and Selection of CRISPR Screening
1. Screening Coverage (Whole Genome or Sub-library)
CRISPR screening can be performed using either whole-genome or sub-libraries. Whole-genome screens design sgRNAs for all coding genes of a species, providing broad coverage but requiring substantial experimental workload. Sub-libraries focus on specific gene groups, such as a particular family, signaling pathway, or functional category.Currently, many pre-designed libraries are available for human, mouse, and green monkey genomes, as well as sub-libraries targeting kinases, nuclear proteins, cyclins, metabolic enzymes, or epigenetic regulators. Library selection should balance coverage with experimental feasibility, ideally choosing the smallest library that meets the screening objectives to reduce workload and costs.
2. Screening Requirements (Loss-of-Function, Gain-of-Function, or Others)
After selecting coverage, the screening strategy must be defined: loss-of-function (LOF) or gain-of-function (GOF).
- Loss-of-function: Typically uses CRISPR-KO or CRISPRi systems. CRISPR-KO generates permanent gene knockout, but for essential or non-coding genes, CRISPRi—a catalytically inactive dCas9 fused to a transcriptional repressor—provides reversible gene knockdown.
- Gain-of-function: Achieved with CRISPRa, which fuses dCas9 to a transcriptional activator to enhance gene expression.
| Type | CRISPR-KO | CRISPRa | CRISPRi |
| Working Principle | Permanent gene knockout via DNA cutting and repair | Transcriptional activation via dCas9-activator | Transcriptional inhibition via dCas9-repressor |
| Pros | ✓ Low background noise ✓ Single-vector system; easy to operate | ✓ No genome breakage ✓ Can target non-coding regions | ✓ No genome breakage ✓ Can regulate transcriptional regions |
| Cons | ✗ Variable editing efficiency ✗ Potential off-targets | ✗ Larger complex; may require stable Cas9 line | ✗ Larger complex; may require stable Cas9 line |
| Applications | Drug resistance/sensitivity screens Synthetic lethality analysis Signaling pathway studies | Gain-of-function analysis Promoter function studies Non-coding region function | Loss-of-function analysis Long non-coding RNA studies |
3. Other Library Considerations
- Barcode libraries: For subtle phenotypes (e.g., metabolism, tissue homeostasis), single-cell sequencing may be required; library backbones can be modified to include barcode sequences.
- Base editing libraries: Use CBE/ABE methods to generate single-nucleotide variant (SNV) libraries targeting specific genes.
4. Screening Purpose (Positive vs. Negative)
- Positive screening: Applies selective pressure so only cells with desired phenotypes survive, enriching key genes.
- Negative screening: Compares sgRNA abundance over time; identifies genes whose loss affects cell survival or function. For example, screening for driver genes or synthetic lethality typically uses negative screening, whereas drug resistance or viral host factor identification uses positive screening.
CRISPR Screening Workflow
Performing a CRISPR screen involves several key steps to systematically study gene function:
1. sgRNA Design and Library Construction
For each gene, design 3–6 sgRNAs and synthesize them using high-throughput methods. Clone the sgRNAs into a lentiviral vector to prepare the screening pool.
2. Lentiviral Packaging and Cell Transduction
Package the sgRNA pool into lentivirus and transduce target cells at low MOI (<0.3), ensuring most cells receive only one sgRNA. The total number of cells should be 200-1000× the number of sgRNAs to maintain library coverage.
3. Cell Screening
Divide the transduced cells into experimental and control groups. Apply screening conditions such as drug treatment or viral infection, and assess phenotypes like proliferation, viability, or resistance.
4. Sequencing and Data Analysis
Extract genomic DNA from both groups, amplify sgRNAs via PCR, and perform NGS sequencing. Analyze sgRNA enrichment or depletion to identify genes linked to the screening phenotype.
5. Functional Validation
Validate candidate genes through individual CRISPR knockout, overexpression, or complementation experiments to confirm their biological role.
Advanced Applications of CRISPR Screening: Target Discovery Strategies Behind a Nature Study
[Useful tips!] How to improve the accuracy of CRISPR library screening targets?
CRISPR Library Screening: Are You Aware of These Details?
CRISPR Screening | A Comprehensive Guide to Building a Functional Screening System
CRISPR Screen Data Analysis
Data analysis is a crucial step in a CRISPR screen, though no single standardized workflow exists. Researchers rely on various bioinformatics tools—such as MAGeCK, MAGeCK-VISPR, ScreenBEAM, BAGEL, CasTLE, ENCoRE, PBNPA, and JACKS—to process and interpret screening results, either individually or in combination.
A typical CRISPR screening analysis workflow includes:
- Quality Control of Sequencing Data
Filter raw NGS reads to remove low-quality or short sequences, producing clean, high-quality data for analysis. - Reads Alignment
Extract sgRNA sequences and align them to the reference sgRNA sequences used in the CRISPR screen. - Statistical Analysis
Count reads, sgRNA coverage, and gene representation to ensure data reliability and detect potential biases. - Differential Analysis Between Samples
Use tools like MAGeCK to compare experimental and control groups, identifying positively or negatively enriched genes linked to the screening phenotype. - Enrichment Analysis
Visualize gene-level results using GSEA (Gene Set Enrichment Analysis) to examine functional pathways through KEGG and GO annotations, aiding interpretation of biological significance.
Applications of CRISPR Screen
What are CRISPR screens used for? In addition to the unique advantages mentioned above, the broad application prospect also contributes to the popularity of CRISPR screen. Here are a few common applications.
1. Cancer treatment
1.1 Identify essential genes of tumor and target oncogenes for treatment
Malignant tumor accumulates many gene mutations, but the maintenance of malignant phenotype of these tumor cells usually depends on only one or some of the oncogenes activated by mutations. This phenomenon is called oncogene addiction, and these oncogenes are also called driver oncogenes. Blocking the activity of these driver oncogenes can induce tumor cells to rapid apoptosis, growth arrest or differentiation, but with little impact on normal cells. This difference brings hope for targeted therapy of tumors.
In recent years, the extensive application of second-generation sequencing has identified many mutation loci in tumor cells, but it is difficult to distinguish which are driver mutations. It is a simpler and more direct method to find out the driving oncogenes that tumor cells depend on for survival by gene inactivation. CRISPR library can simultaneously target different genes throughout the genome, which is an excellent tool to achieve high-throughput gene inactivation. In 2014, Shalem et al. [1] first identified the key genes for the survival of human melanoma cells and pluripotent stem cells using the GeCKO Library (Fig. 1). Wang et al. [2] identified NCAPG as an essential oncogene for hepatocellular carcinoma tumor growth by whole genome crispr screen. Kiessling et al. [3] verified the driving genes EGFR of HCC-827 cell line and NRAS and MEK1 of CHP-212 cell line by using whole genome CRISPR library screening, and found new driving genes TBK1 and TRIB2 for further research.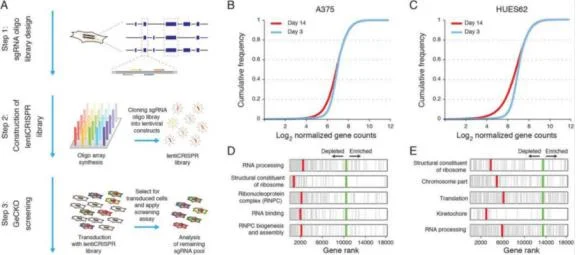
View Picture
Figure 1. Screening A375 and HUES62 survival essential genes by CRISPR library
1.2 Screen synthetic lethality genes to achieve precise treatment
For oncogenes, inhibitors can be used for targeted therapy, but for tumor suppressor genes, the inactivation of their functions makes it impossible to directly target them. So the synthetic lethality gene of the suppressor gene is usually targeted and inactivated. Synthetic lethality refers to the phenomenon that two non-lethal mutant genes inactivate at the same time leading to cell death. If there is inactivation of gene A in tumor cells, drugs can be used to inhibit its synthetic lethality gene B. By inactivate both genes in tumor cells, cell death would occur. However, healthy somatic cells can still have normal physiological functions because they have normal gene A. Thus, the drug specifically kills tumor cells (see Fig. 2) [4].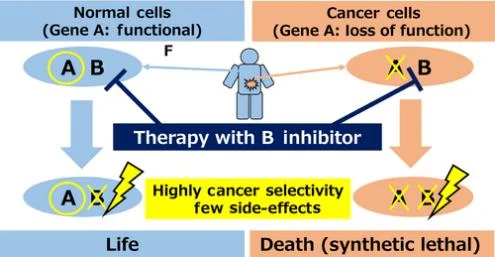
View Picture
Figure 2. Synthetic lethality therapy
The most typical application of synthetic lethality is the treatment of BRCA1/2 mutated tumors with PARP inhibitors. By 2020, the global sales of PARP inhibitors had exceeded 2 billion US dollars. So how to find more gene pairs with synthetic lethality? CRISPR library high-throughput screening plays a big role! CRISPR library screening for synthetic lethality generally has two logics. One is to directly design a dual-gRNA vector, that is, to simultaneously target two genes in one cell and screen gene pairs with synthetic lethality. Shen et al. [5] used this method to screen 73 genes in three cancer cell lines (Hela, A549 and 293T) by 150000 combinations and found 120 gene pairs with synthetic lethality (see Fig. 3). Another logic is to screen a library on a single tumor suppressor gene knockout cell line to screen out targets that have synthetic lethal effects with the knockout gene. Feng et al.[6] used this method to first construct 12 known tumor suppressor gene knockout cell lines on 293A, and then screen wild-type and knockout cell lines with a genome-wide knockout library to analyze the changes in gRNA abundance before and after cell screening. Potential synthetic lethal genes were identified by comparing the abundance differences of gRNAs between wild-type and knockout cell lines after CRISPR library screening.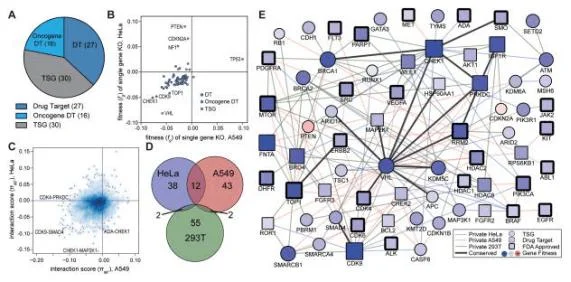
View Picture
Figure 3. Screening gene interaction relationships using CRISPR library
1.3 Search for tumor drug resistance related genes, study drug resistance mechanisms, and optimize treatment strategies
The main problem of tumor molecular targeted drugs is drug resistance, so it is of great clinical significance to explore the mechanism of drug resistance. Through positive screening of CRISPR library, most cells died due to drug sensitivity, and some cells survived due to drug resistance by knockout specific gene. The genes related to drug resistance can be screened out by detecting the gRNAs carried by the surviving cells.
Vemurafenib is approved for the treatment of advanced melanoma carrying the BRAFV600E mutation. In order to study the drug resistance mechanism of Vemurafenib, Shalem et al. [1] designed a knockout library containing 64751 sgRNAs and targeting 18080 genes in the whole genome. After cell infection and expansion, nf1, med12, nf2, cul3, tada2b, tada1 and other drug resistance related genes were screened out. A new hypothesis of the mechanism of Vemurafenib tumor resistance was proposed.
2. Screening host factors related to viral infection
CRISPR library screening is often used to study host factors related to viral infection. For hepatitis C virus, dengue virus, Japanese encephalitis virus and West Nile virus, a number of host factors related to endoplasmic reticulum function and receptors have been screened out. For example, Zhang et al. [7] verified nine human genes required for flavivirus infection through genome-wide CRISPR/Cas9 screening, all of which are related to endoplasmic reticulum (ER) functions (including translocation, protein degradation and N-linked glycosylation). Ma et al. [8] screened the whole genome by designing a library containing 77406 sgRNAs and targeting 20121 genes, and then obtained seven genes belong to the endoplasmic reticulum related protein degradation pathway (ERAD), such as EMC2, EMC3 and SEL1L. Further experiments showed that knockdown of these genes would not prevent the replication of West Nile virus (WNV), but would prevent the virus from killing cells.
More CRISPR screen applications in the field of virus
>>>CRISPR Screening Library: Opening a New Perspective for Virus Research - Special Guide>>>New Approach to Screening Viral Restriction Factors Using CRISPR Screening Libraries!
3. Screening of antibody target
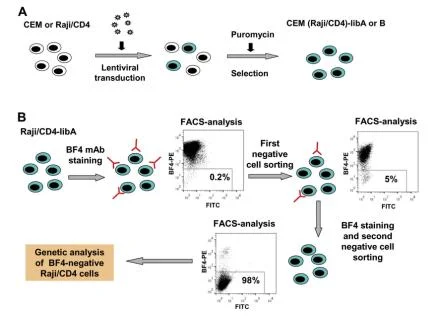
View Picture
Figure 4. Verification of BF4 antibody specificity using CRISPR library
4. Study of signal pathway
Pyroptosis is an immune defense reaction initiated by the body after sensing the infection of pathogenic microorganisms. Inflammation activated caspase-1 and caspase-4, caspase-5 and caspase-11, which recognize bacterial lipopolysaccharides, can cause cell pyroptosis, but the mechanism remains unknown. Shi et al. [10] first established a lipopolysaccharide (LPS) electrotransformation method that can induce more than 90% cell pyroptosis, and then transduced a CRISPR knockout library into Tlr4-/- bone marrow-derived immortalized macrophages (iBMDMs) that can normally respond to lipopolysaccharide stimulation, and sequenced and analyzed the cells that survived after lipopolysaccharide induced cell pyroptosis. The analysis results showed that among the five sgRNAs targeting the gasdermin D (GSDMD) gene, four had copy numbers in the top 30, of which two were in the top 10. The subsequent results also further proved that the N-terminal of GSDMD could induce cell pyroptosis. To sum up, the researchers used CRISPR gene library to carry out genome-wide genetic screening, successfully screened the gene GSDMD that can inhibit cell pyroptosis after knockout, and clarified the molecular mechanism of GSDMD as an inflammatory caspase substrate protein that can trigger cell pyroptosis after being cleaved.

View Picture
Figure 5. LFn-Bsak CRISPR-Cas9 screen hits with multiple gRNAs
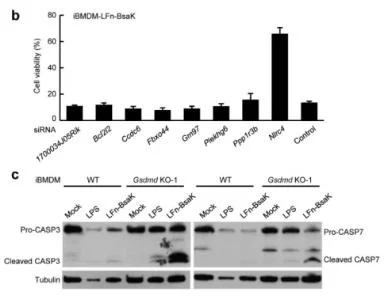
View Picture
Figure 6. siRNA knockdown validation of screen hits
Challenges and Suggestions
High-throughput whole-genome CRISPR screening holds tremendous promise for functional genomics, yet several practical challenges remain.
Main Challenges
1. Off-Target Effects
Ensuring high target specificity is critical for reliable results, but off-target activity can interfere with accuracy. Additionally, gain- or loss-of-function screens can be influenced by cell heterogeneity, which may cause preferential changes in gene expression and complicate interpretation.
2. Limitations of Delivery Systems
Current delivery vectors often limit applications due to efficiency or immunogenicity in primary cells. In vivo screens face additional challenges from scalability and tissue accessibility, emphasizing the need for more efficient and safe delivery methods.
3.Complexity of Multi-Factor Interactions
Studying cooperative gene networks is challenging. Designing thousands of sgRNA combinations for double or multiple gene knockouts is labor-intensive, and integrating multi-factor data remains difficult due to limited analytical tools.
Optimization Directions
1. Enhance Library and Delivery Technology
Incorporate barcoded or inducible libraries and single-cell sequencing to trace lineage and account for cell heterogeneity. Develop advanced delivery systems leveraging spatial transcriptomics to enable functional identification of key regulators in vivo.
2. Optimize sgRNA Design and Analytical Tools
Target conserved domains to reduce the number of sgRNAs needed for complex screens. Employ AI or deep learning to model gene interaction networks and uncover cooperative regulatory mechanisms.
3. Improve Data Resources and Interdisciplinary Integration
Establish standardized databases and analytical pipelines to facilitate multi-omics data integration, supporting mechanistic interpretation of complex phenotypes.
Summary and Prospect
As a key tool in functional genomics, CRISPR screening has driven major advances in both basic research and translational medicine. Further development depends on progress in several areas:
01.
Technological Innovation
Develop delivery systems that balance efficiency and safety—such as integrated in vivo spatial-genomics platforms—to overcome barriers in primary cells and live models.
02.
Methodological Upgrades
Improve multi-factor screening and data analysis through the integration of computational biology and experimental science, enabling a deeper understanding of complex regulatory networks.
03.
Clinical Translation
Integrate CRISPR screen results with disease models to accelerate the translation from research to precision medicine, such as designing personalized therapeutic strategies.
With advances in multi-omics, artificial intelligence, and delivery technologies, CRISPR screening will increasingly reveal the molecular mechanisms that govern life processes, offering unprecedented insight for research and therapy.
Considerations for Selecting a CRISPR Screening Platform
When choosing a CRISPR screening platform, researchers should evaluate:
- ✓ Library quality and coverage – High transformation efficiency and uniform sgRNA distribution are essential for reproducible results.
- ✓ Experimental flexibility – Support for diverse screening conditions, including drug treatment and infection models.
- ✓ Data analysis capability – Robust statistical algorithms and intuitive visualization tools to interpret large-scale screening data.
- ✓ Resource availability – Access to ready-to-use plasmids, viral particles, or engineered cell pools to accelerate project initiation.
FAQs
1. What are CRISPR screens used for?
CRISPR screens are used to identify genes that influence specific biological processes or phenotypes by systematically knocking out or editing genes across a cell population. They are particularly useful for:
- Identifying Essential Genes: Discovering genes required for cell survival or growth.
- Finding Drug Targets: Identifying genes whose knockout confers resistance to a drug, indicating potential therapeutic targets.
- Understanding Gene Function: Elucidating the roles of specific genes in biological pathways or responses.
- Studying Disease Mechanisms: Identifying genes involved in disease processes, such as cancer or neurodegenerative disorders.
- Exploring Genetic Interactions: Understanding how genes interact in complex networks to influence cellular behavior.
CRISPR screens are a powerful tool for functional genomics and can accelerate the discovery of new biological insights and potential therapeutic strategies.
2. How to design a CRISPR screen?
Designing a CRISPR screen involves careful planning and consideration of several factors to ensure robust and reliable results. Here’s a step-by-step guide to designing a CRISPR screen:
- Define the Objective:
- Objective: Clearly define the biological question or phenotype you want to study (e.g., identifying genes involved in drug resistance or essential for cell survival).
- Screen Type: Decide whether you need a positive screen (identifying genes that confer resistance) or a negative screen (identifying essential genes).
- Choose the Right CRISPR Library
- Library Type: Select a pre-designed CRISPR library targeting the genes of interest or a genome-wide library for broader screening.
- sgRNA Design: Ensure the library includes multiple sgRNAs per gene to increase the likelihood of effective gene knockout.
- Optimize Delivery Methods
- Delivery Method: Choose an appropriate delivery method for CRISPR components (e.g., viral vectors like AAV or lentivirus, electroporation, or lipid nanoparticles).
- Transduction Efficiency: Optimize transduction conditions to achieve high efficiency and uniform coverage of sgRNAs.
- Establish Experimental Conditions
- Cell Line: Select a suitable cell line that is relevant to your biological question and amenable to CRISPR editing.
- Selective Pressure: Define the conditions under which the screen will be performed (e.g., drug treatment, environmental stress).
- Time Course: Determine the duration of the experiment based on the expected time for phenotypic changes to occur.
- Ensure Sufficient Coverage
- Coverage: Aim for a coverage of at least 500-1,000 cells per sgRNA to ensure robust representation and statistical power.
- Initial Cell Number: Calculate the total number of cells needed based on the library size and desired coverage.
3. What is the coverage of the CRISPR screening?
The coverage of a CRISPR screen refers to the number of cells transduced with each sgRNA in the library. It is a critical parameter that affects the reliability and reproducibility of the screening results. Here are the key points regarding CRISPR screening coverage:
- Coverage Definition: represents the number of cells transduced with each sgRNA. For example, a coverage of 500 means that each sgRNA is present in 500 cells on average.
- Recommended Coverage:
- Genome-Wide Screens: Typically require a coverage of 500-1,000 cells per sgRNA.
- Small-Scale Screens: For screens targeting a smaller number of genes, lower coverage (e.g., 300-500 cells per sgRNA) may be sufficient.
The coverage of CRISPR screening is crucial for ensuring the reliability and reproducibility of the results. Genome-wide screens typically require a coverage of 500-1,000 cells per sgRNA, while smaller screens may need less. High coverage helps in achieving robust results and overcoming experimental noise.
4. How to validate a CRISPR screen?
- Replication: Repeat the screen independently. Consistent results across replicates validate identified targets.
- Controls: Include positive and negative control genes. Positive controls should show expected phenotypes, and negative controls should not.
- Phenotype Confirmation: Use alternative methods to verify phenotypes related to identified genes, like cell proliferation assays.
- Rescue Experiments: Re - express knocked - out genes. Phenotype reversal validates the gene - phenotype link.
- Bioinformatics: Analyze for off - target effects and perform pathway analysis.
- Literature Comparison: Compare results with previous studies. Consistent findings add credibility; novel ones need further investigation.
5. How many cells for CRISPR screen?
The number of cells required for a CRISPR screen depends on several factors, including the complexity of the library, the desired coverage, and the specific experimental design. Here are some general guidelines based on recent studies and best practices:
General Recommendations:
Genome-Wide Screens:
- Coverage: Aim for a coverage of 500-1,000 cells per sgRNA to ensure robust representation and statistical power.
- Total Cell Number: For a typical genome-wide CRISPR library with 10,000-20,000 sgRNAs, you would need at least 5-10 million cells to achieve a coverage of 500-1,000 cells per sgRNA.
Small-Scale Screens:
- Coverage: For screens targeting a smaller number of genes (e.g., 1,000 sgRNAs), a coverage of 300-500 cells per sgRNA may be sufficient.
- Total Cell Number: This would require approximately 300,000-500,000 cells.
Example Calculation:
- Library Size: 15,000 sgRNAs
- Desired Coverage: 500 cells per sgRNA
- Desired Coverage: 500 cells per sgRNA
Reference
[1] Shalem O, Sanjana NE, Hartenian E, Shi X, Scott DA, Mikkelson T, Heckl D, Ebert BL, Root DE, Doench JG, Zhang F. Genome-scale CRISPR-Cas9 knockout screening in human cells. Science. 2014 Jan 3;343(6166):84-87.[2] Wang Y, Gao B, Tan PY, Handoko YA, Sekar K, Deivasigamani A, Seshachalam VP, OuYang HY, Shi M, Xie C, Goh BKP, Ooi LL, Man Hui K. Genome-wide CRISPR knockout screens identify NCAPG as an essential oncogene for hepatocellular carcinoma tumor growth. FASEB J. 2019 Aug;33(8):8759-8770.
[3] Kiessling M K, Schuierer S, Stertz S, et al. Identification of oncogenic driver mutations by genome-wide CRISPR-Cas9 dropout screening[J]. BMC genomics, 2016, 17(1): 1-16.
[4] Sasaki M, Ogiwara H. Synthetic lethal therapy based on targeting the vulnerability of SWI/SNF chromatin remodeling complex‐deficient cancers[J]. Cancer Science, 2020, 111(3): 774-782.
[5] Shen JP, Zhao D, Sasik R, Luebeck J, Birmingham A, Bojorquez-Gomez A, Licon K, Klepper K, Pekin D, Beckett AN, Sanchez KS, Thomas A, Kuo CC, Du D, Roguev A, Lewis NE, Chang AN, Kreisberg JF, Krogan N, Qi L, Ideker T, Mali P. Combinatorial CRISPR-Cas9 screens for de novo mapping of genetic interactions. Nat Methods. 2017 Jun;14(6):573-576.
[6] Feng X, Tang M, Dede M, et al. Genome-wide CRISPR screens using isogenic cells reveal vulnerabilities conferred by loss of tumor suppressors[J]. Science advances, 2022, 8(19): eabm6638.
[7] Zhang R, Miner JJ, Gorman MJ, Rausch K, Ramage H, White JP, Zuiani A, Zhang P, Fernandez E, Zhang Q, Dowd KA, Pierson TC, Cherry S, Diamond MS. A CRISPR screen defines a signal peptide processing pathway required by flaviviruses. Nature. 2016 Jul 7;535(7610):164-8.
[8] Ma H, Dang Y, Wu Y, Jia G, Anaya E, Zhang J, Abraham S, Choi JG, Shi G, Qi L, Manjunath N, Wu H. A CRISPR-Based Screen Identifies Genes Essential for West-Nile-Virus-Induced Cell Death. Cell Rep. 2015 Jul 28;12(4):673-83.
[9] Zotova A, Zotov I, Filatov A, Mazurov D. Determining antigen specificity of a monoclonal antibody using genome-scale CRISPR-Cas9 knockout library. J Immunol Methods. 2016 Dec;439:8-14.
[10] Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, Zhuang Y, Cai T, Wang F, Shao F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015 Oct 29;526(7575):660-5.
 Promotions
Promotions 








