Monofunctional Inhibitors Trigger CRISPR-Validated Kinase Degradation


Introduction
Targeted protein degradation (TPD) technologies—including PROTACs and molecular glues—typically rely on bifunctional molecules that induce proximity between a target protein and an E3 ubiquitin ligase. While powerful, these approaches often face drug-likeness limitations, complex pharmacokinetics, and increased risk of off-target effects. In a landmark study published in Nature, the research team led by Georg E. Winter at the Austrian Academy of Sciences demonstrated that monofunctional kinase inhibitors can selectively induce target degradation by “supercharging” endogenous proteolytic circuits. With CRISPR library screening as the central enabling technology, the researchers systematically mapped the degradation pathways triggered by kinase inhibitors. Through these screens, they pinpointed the key regulatory components governing degradation and elucidated distinct mechanisms underlying the turnover of three kinases—LYN, BLK, and RIPK2. This work challenges the prevailing paradigm that TPD requires bifunctional degrader molecules, establishing a new conceptual framework for monofunctional degrader discovery. Throughout the study, CRISPR pooled library screening served as the core mechanistic discovery engine, enabling the precise dissection of endogenous degradation pathways and providing critical insights for next-generation targeted degradation therapeutics.

Research Background
Kinase inhibitors represent one of the most widely used classes of targeted therapeutics in clinical practice. Interestingly, several inhibitors have been observed to induce degradation of their target kinases, a phenomenon commonly attributed to disruption of interactions with the molecular chaperone HSP90. However, the prevalence, generality, and underlying mechanisms of this effect remain incompletely understood. At the same time, bifunctional degraders, such as PROTACs, have demonstrated considerable therapeutic potential but face notable challenges, including limited membrane permeability, poor oral bioavailability, and complex chemical synthesis. Consequently, elucidating the principles and mechanisms by which monofunctional small molecules induce target protein degradation could provide critical insights for the development of next-generation targeted degradation therapeutics.
Research Objectives
Systematically identify monofunctional kinase inhibitors with degradation-inducing activity, and determine the scope, selectivity, and prevalence of this phenomenon. Elucidate the molecular mechanisms underlying inhibitor-induced kinase degradation, moving beyond the traditional “molecular chaperone deprivation” model. Establish a conceptual framework of “supercharging endogenous degradation pathways”, providing mechanistic guidance for the development of monofunctional degrader therapeutics.
Research Methods
- Cell Line Construction: To investigate degradation mechanisms for the three target kinases—LYN, BLK, and RIPK2—the researchers generated protein stability reporter cell lines by fusing each kinase to a BFP–P2A–mCherry dual-fluorescent reporter in either KBM7 iCas9 or RKO iCas9 backgrounds. This design enabled precise quantification of protein stability and degradation using flow cytometry.
-
CRISPR Library Selection and Construction
- LYN degradation mechanism study: A ubiquitin–proteasome system (UPS)-focused sgRNA library containing 7,801 sgRNAs was used to specifically interrogate genes associated with protein degradation pathways.
- BLK and RIPK2 mechanism studies: A genome-wide sgRNA library was applied to enable unbiased identification of all potential regulatory genes.
Lentiviral transduction was performed at a multiplicity of infection (MOI) of 0.1–0.2, ensuring that each cell integrated a single sgRNA. After transduction, cells were selected with G418 (1 mg/mL) for 14 days to generate a stable pooled knockout library, maintaining ~1000× library coverage.
- Multi-Omics Validation: Multiple orthogonal approaches were used to validate screening results and identify degradation-associated pathways: Quantitative proteomics, Chemical proteomics (Kinobead profiling), and Proximity labeling (BioID). These techniques enabled the identification of protein interactors and signaling pathways associated with kinase degradation.
- Functional Validation: Mechanistic validation was performed through: CRISPR–Cas9 gene knockout, Drug rescue experiments, Co-immunoprecipitation (Co-IP), and Immunofluorescence imaging. Protein stability and degradation efficiency were quantified using flow cytometry and Western blotting.
- Structural Biology Analysis: To investigate the structural basis of degradation mechanisms, the study used AlphaFold3 to predict the structure of the BLK–γ-secretase complex, followed by molecular dynamics simulations to analyze potential interaction interfaces and regulatory mechanisms.
- Mechanistic Model Systems: Three representative kinases were selected to illustrate distinct degradation pathways: LYN – strongly dependent on HSP90, BLK – weakly dependent on HSP90, and RIPK2 – independent of HSP90. These models enabled systematic dissection of divergent kinase degradation mechanisms.
Research Workflow
- High-Throughput Screening: The researchers constructed a kinase inhibitor–target degradation landscape by profiling interactions between kinase inhibitors and kinase stability. This analysis identified specific degradation pairs and assessed correlations between degradation susceptibility and: HSP90 client status, degradation potential in PROTAC systems.
- Mechanistic Classification: LYN model: revealed a degradation mechanism mediated by an “activity–stability switch.” BLK model: uncovered a γ-secretase–dependent membrane localization regulatory mechanism controlling kinase turnover. RIPK2 model: demonstrated higher-order assembly–induced autophagic degradation.
- Conceptual Framework Development: The study ultimately established a general framework in which small-molecule inhibitors drive targets into a “degradation-prone state” by superactivating endogenous proteolytic pathways, providing a conceptual basis for monofunctional degrader drug discovery.
Key Findings
1. Inhibitor-Induced Kinase Degradation Is a Widespread Yet Selective Phenomenon
Dynamic interaction profiling across 98 kinases (88 wild-type and 10 mutant variants) and 1,570 kinase inhibitors identified: 232 inhibitors capable of inducing degradation and 160 specific kinase–inhibitor degradation pairs. Among the tested kinases, HER2, ABL1, and RAF1 (S257L) were the most degradation-prone targets. Degradation activity showed a strong correlation with HSP90 client status: strong and weak HSP90 client kinases exhibited significantly higher degradation rates than non-clients. However, approximately 40% of degradation events could not be explained by the classical “chaperone deprivation” model, indicating additional mechanisms. Notably, inhibitor-induced degradation showed no correlation with PROTAC degradation profiles, suggesting that these mechanisms are fundamentally distinct. Furthermore, mutant kinases exhibited markedly different degradation patterns compared with their wild-type counterparts (Jaccard distance ≥ 0.8), indicating that mutations can substantially alter degradation susceptibility. Based on the screening results, the researchers selected three representative kinases— LYN, BLK, and RIPK2 —each displaying clear and highly specific degradation trajectories, to serve as core model systems for subsequent mechanistic investigations.
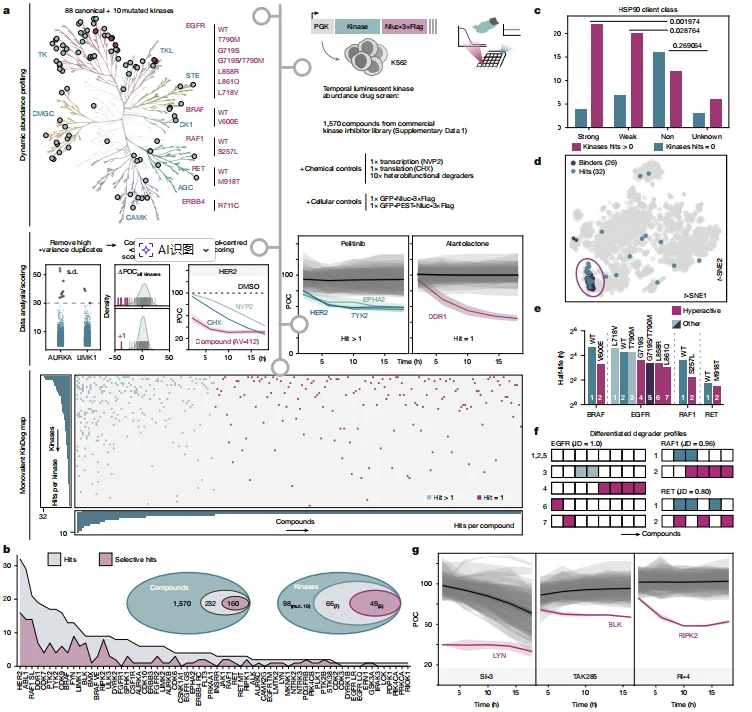
Figure 1. Kinase Degradation (KinDeg) Landscape of 1,570 Monovalent Kinase-Targeting Compounds
2. Mechanism of LYN Degradation: Inhibition of an Upstream Regulatory Kinase Activates an “Activity–Stability Switch”
The inhibitor SI-3 induces degradation of LYN by selectively inhibiting the upstream regulatory kinase CSK, thereby relieving its negative regulatory control over LYN. This leads to LYN activation and phosphorylation at residues Y32 and Y316, generating a dual-phosphorylated phosphodegron that triggers protein degradation. CRISPR screening using a UPS-focused sgRNA library identified the E3 ubiquitin ligase CBL as the most significantly enriched hit (log₂[FC] > 1.585, P < 0.05). Its homolog CBLB was also strongly enriched, suggesting functional redundancy between the two ligases. CBL is the most significantly enriched hit Functional validation experiments demonstrated that:
- Single knockout of either CBL or CBLB did not block SI-3–induced LYN degradation.
- Double knockout of CBL and CBLB completely abolished the degradation phenotype, confirming their cooperative function.
Further mechanistic analysis revealed distinct degradation routes mediated by the two ligases:
- CBL drives LYN degradation through both proteasomal and lysosomal pathways.
- CBLB mediates degradation exclusively via the proteasome pathway.
Chemical rescue experiments further supported this model. Inhibitor treatment demonstrated that LYN degradation is dependent on ubiquitination, as it can be blocked by the kinase inhibitor TAK285. Moreover, degradation required both proteasomal and lysosomal pathways, consistent with the dual degradation mechanisms mediated by CBL and CBLB. Protein interaction analyses showed that SI-3 treatment markedly enhances the interaction between CBL/CBLB and LYN, facilitating ubiquitination and subsequent turnover.
Biochemical profiling revealed that SI-3 preferentially inhibits CSK with a dissociation constant of K_D = 34.60 nM, thereby removing CSK-mediated suppression of LYN. This results in activation of LYN and phosphorylation at residues Y32 and Y316, generating the dual-phosphorylation phosphodegron required for degradation. Importantly, the double mutant LYN (Y32A/Y316A) is completely resistant to SI-3–induced degradation, confirming that phosphorylation at these sites is essential for triggering the degradation process.
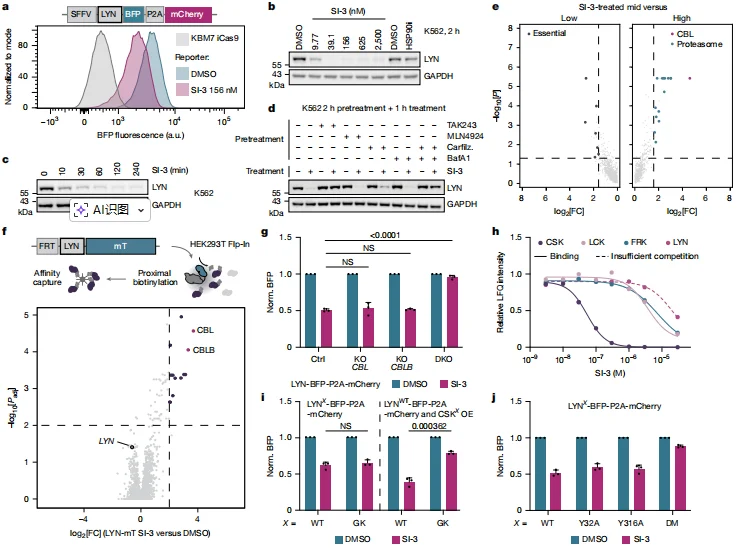
Figure 2. SI-3 Rapidly Induces LYN Degradation Through a Classical Activity–Stability Switch Mechanism
3. Mechanism of BLK Degradation: γ-Secretase–Mediated Membrane Relocalization Triggers Cytosolic Degradation
Following genome-wide CRISPR library screening, all four subunits of the γ-secretase complex were identified as high-confidence enriched hits, including APH1A, NCSTN, PSEN1, and PSENEN. This finding indicates that degradation of BLK is strictly dependent on γ-secretase activity. Pharmacological and genetic validation experiments further confirmed this mechanism:
- Pharmacological inhibition of γ-secretase using DAPT pretreatment completely blocked TAK285-induced BLK degradation.
- Genetic knockout of PSENEN likewise abolished BLK degradation triggered by TAK285.
- In contrast, BLK degradation induced by HSP90 inhibition remained unaffected, demonstrating that γ-secretase specifically mediates the TAK285-driven degradation pathway.
Mechanistically, TAK285 activates γ-secretase through a network-mediated effect, leading to proteolytic cleavage of the N-terminal myristoylation motif of BLK. This cleavage disrupts membrane anchoring and causes BLK to translocate from the plasma membrane to the cytosol, where it subsequently undergoes degradation.
Detailed sequence analysis revealed that the first seven amino acids at the BLK N-terminus—including residues G2, L3, V4, and S6—constitute a critical degradation motif. Substituting this motif into the related Src-family kinase SRC was sufficient to confer TAK285-induced degradation sensitivity, highlighting its functional importance. Structural modeling using AlphaFold3 further supported this mechanism. The predictions indicate that the BLK N-terminal domain inserts into the catalytic site of the γ-secretase complex, with the L3 residue positioned within a hydrophobic pocket, providing a structural basis for the sequence-specific degradation determinant.
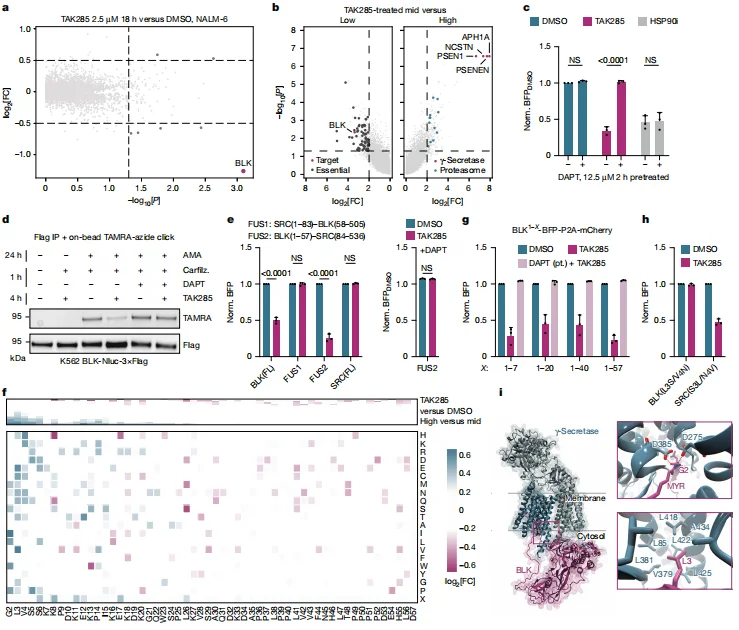
Figure 3. BLK Is Degraded by TAK285 in a γ-Secretase–Dependent Manner
4. Mechanism of RIPK2 Degradation: Induction of Higher-Order Assemblies Triggers Autophagic Turnover
Genome-wide CRISPR sgRNA library screening identified several key regulators of autophagy and ubiquitin signaling as significantly enriched hits, including the essential autophagy factor FIP200, the ubiquitin-like protein TMUB1, and the E3 ubiquitin ligases BIRC2 (cIAP1) and XIAP. Gene ontology analysis further revealed that the enriched genes were strongly associated with lysosome-mediated degradation pathways, suggesting a central role for autophagy in this process. Functional validation experiments confirmed this mechanism:
- Knockout of the essential autophagy gene FIP200 or pharmacological inhibition of lysosomal acidification using Bafilomycin A1 significantly blocked RI-4–induced degradation of RIPK2.
- In contrast, knockout of PSMB5 had no effect on degradation, demonstrating that RIPK2 turnover is mediated by macroautophagy rather than the proteasome.
Mechanistic investigation showed that TMUB1 plays a key role in the formation of RIPK2 higher-order assemblies, known as RIPosomes. Loss of TMUB1 significantly delayed RIPosome formation, thereby attenuating RIPK2 degradation. The small molecule RI-4 promotes CARD-domain–dependent oligomerization of RIPK2, which drives the formation of these higher-order assemblies. The resulting complexes recruit autophagy adaptor proteins such as SQSTM1 (p62), thereby initiating selective autophagic degradation. Ubiquitination was also shown to be essential for efficient clearance of these assemblies. Double knockout of BIRC2 and XIAP, or introduction of the RIPK2 I212D mutation, which disrupts binding to IAP family ligases, impairs the removal of RIPK2 assemblies, confirming that ubiquitin signaling is a critical determinant of assembly turnover.
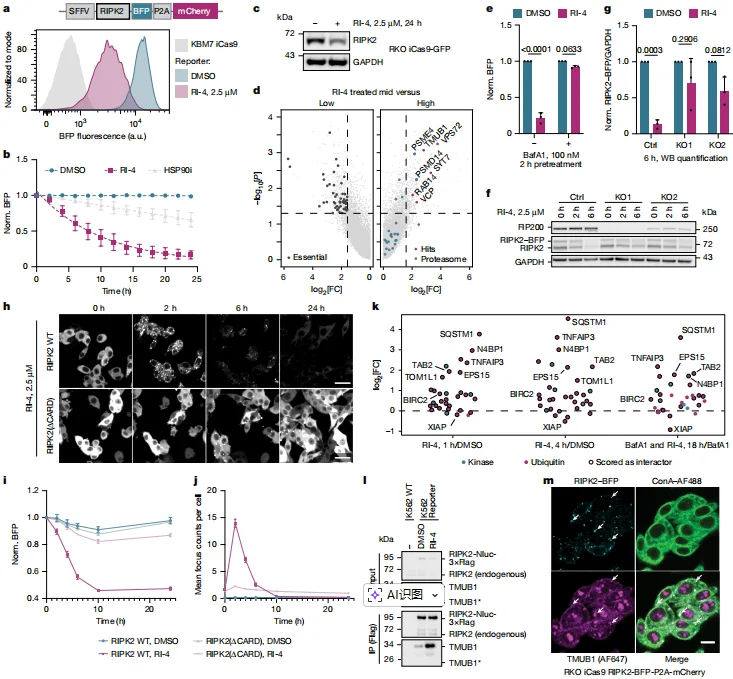
Figure 4. RI-4 Destabilizes RIPK2 Through TMUB1-Mediated Oligomerization and Macroautophagy
Summary
Through systematic screening and mechanistic dissection, this study demonstrates that monofunctional kinase inhibitors can selectively induce degradation of their targets by driving proteins into a “degradation-prone state” and activating endogenous proteolytic pathways. The work establishes both the prevalence and selectivity of inhibitor-induced kinase degradation and elucidates three representative mechanistic paradigms:
- Activity regulation–driven degradation, exemplified by LYN.
- Subcellular localization–dependent degradation, illustrated by BLK.
- Higher-order assembly–triggered degradation, demonstrated by RIPK2.
Together, these findings establish the conceptual framework of “supercharging endogenous degradation pathways.” This discovery significantly expands our understanding of small molecule–target protein interactions and introduces a new paradigm for targeted protein degradation drug discovery that does not require bifunctional degrader design. As such, it holds substantial promise for translational applications in next-generation therapeutic development.
Ubigene CRISPR-iScreen™ Libraries
CRISPR-iScreen™ is an innovative CRISPR screening technology independently developed by Ubigene, designed to enable high-efficiency pooled CRISPR screening. Ubigene currently offers more than 40 ready-to-use CRISPR libraries, covering multiple species, including: human, mouse, cynomolgus monkey, and pig. These libraries support genome-wide CRISPR knockout (KO), CRISPR interference (CRISPRi), and CRISPR activation (CRISPRa) screening approaches. In addition to genome-wide libraries, Ubigene provides focused sub-libraries targeting specific biological pathways and gene families, including: Kinases, Cell cycle regulators, Membrane proteins, and Metabolic pathways. These resources enable researchers to rapidly perform target discovery and functional screening with greater precision and efficiency. Currently, Ubigene maintains an extensive inventory of ready-to-ship screening reagents, including: l 150+ CRISPR library virus stocks l 400+ CRISPR library cell pools l 40+ CRISPR library plasmids stocks In addition, Ubigene offers end-to-end CRISPR screening services, supporting both in vitro and in vivo functional screening, ensuring seamless support for your research from library design to data interpretation.
Contact us to learn more>>>Reference
Scholes NS, Bertoni M, Comajuncosa-Creus A, Kladnik K, Guo X, Frommelt F, Hinterndorfer M, Razumkov H, Prokofeva P, Schwalm MP, Born F, Roehm S, Imrichova H, Santini BL, Barone E, Schätz C, Muñoz I Ordoño M, Lechner S, Rukavina A, Serrano I, Abele M, Koren A, Kubicek S, Knapp S, Gray NS, Superti-Furga G, Kuster B, Shi Y, Aloy P, Winter GE. Inhibitors supercharge kinase turnover through native proteolytic circuits. Nature. 2026 Jan;649(8098):1032-1041. doi: 10.1038/s41586-025-09763-9. Epub 2025 Nov 26. PMID: 41299171; PMCID: PMC12823440.
 Promotions
Promotions 








