Research Frontier | Fighting Cancer with Cancer? Cystatin C Activates the TREM2 Pathway, Opening a New Therapeutic Avenue for Alzheimer’s Disease


Introduction
Alzheimer's disease (AD) and Cancer are among the most devastating diseases worldwide. Intriguingly, epidemiological studies have long observed that individuals with a history of cancer exhibit a significantly lower incidence of Alzheimer’s disease. Despite this inverse correlation, the biological mechanisms underlying this interaction have remained largely unclear. In a recent study published in Cell, a research team from Huazhong University of Science and Technology reported a groundbreaking discovery: peripheral tumors can secrete Cystatin C (Cyst-C), which binds to amyloid-β oligomers and activates the microglial receptor TREM2. Activation of this pathway enhances microglia-mediated clearance of established amyloid plaques, ultimately reducing neuropathological damage and improving cognitive function in AD mouse models. This finding not only introduces a new perspective at the intersection of cancer biology and neuroscience, but also breaks through the limitations of traditional amyloid-reduction strategies, offering an innovative conceptual framework for precision therapeutic development in Alzheimer’s disease.

Research Background
Alzheimer's disease (AD) is a progressive neurodegenerative disorder characterized by gradual cognitive decline. One of its hallmark pathological features is the accumulation of β-amyloid (Aβ) plaques, which form extracellular senile plaques in the brain. Although several anti-Aβ monoclonal antibodies have recently received clinical approval, their long-term efficacy and safety remain controversial, and effective disease-modifying therapies capable of clearing pre-existing plaques are still lacking. Interestingly, Cancer and AD share multiple risk factors, including smoking exposure and dysregulation of cell cycle control. Epidemiological studies have consistently reported a negative correlation between the incidence of cancer and AD. However, confounding factors such as differences in patient survival time and diagnostic bias have complicated interpretation. As a result, the direct impact of peripheral tumors on AD pathology and the underlying molecular mechanisms have remained largely unresolved.
Research Objectives
This study aimed to: l Determine whether peripheral cancers influence amyloid pathology in AD. l Identify key tumor-secreted effector molecules responsible for this effect. l Elucidate the signaling pathways mediating cancer-associated improvement in AD pathology. Ultimately, the goal was to provide experimental evidence for the development of novel therapeutic strategies targeting Alzheimer’s disease.
Research Methods
- Animal Model Construction: Tumor-bearing AD mouse models were established by intravenous injection of cancer cells into the tail vein of transgenic AD models: l 3-month-old 5xFAD mouse model mice, representing an amyloid pathology model. l 5-month-old rTg4510 mouse model mice, representing a tau pathology model. The following tumor cell lines were used: l Lewis lung carcinoma (LLC) l RM1 prostate cancer cell line l MC38 colon adenocarcinoma cell line To verify pathway specificity, additional genetically modified mouse models were generated, including: l Microglia-specific TREM2 knockout mice (Cx3cr1-TREM2−/−) l TREM2R47H mutation model l Cystatin C L68Q mutation model
- Knockout Cell Line Construction: Using CRISPR-Cas9 genome editing, the researchers generated gene knockout LLC tumor cell lines targeting several candidate secreted factors, including: CLS1, CPE, Cst3 (encoding Cystatin C), and GSN. These engineered cell lines were used to identify and validate tumor-derived effector molecules responsible for modulating AD pathology.
- Intervention and Treatment: Cancer cell–secreted proteins (CSPs) were isolated and administered via tail vein injection to AD mice. Animals received continuous treatment for 30–60 days to assess therapeutic effects.
- Behavioral Assessment: Cognitive function was evaluated using established behavioral assays: Morris water maze for spatial learning and memory, and Y-maze spontaneous alternation test for working memory.
- Neuropathological Analysis: Amyloid pathology was assessed using: Immunofluorescence staining for Aβ1–16, Thioflavin S staining to visualize amyloid plaques, and Western blotting to quantify plaque burden and related protein expression.
- Molecular Mechanism Validation: Multiple molecular approaches were applied to dissect the underlying mechanisms: l RNA sequencing (RNA-seq) and proteomics to identify differentially expressed molecules l ELISA to quantify Cystatin C levels in plasma and cerebrospinal fluid l FRET assays to validate interactions between TREM2 and DAP12 l Flow cytometry to analyze immune cell subsets l ¹²⁵I radiolabeling experiments to evaluate the blood–brain barrier permeability of Cystatin C
- Clinical Sample Validation: To confirm clinical relevance, plasma samples from patients with multiple cancers—including: Non-small cell lung cancer, Prostate cancer, and Colorectal cancer. These were analyzed to measure circulating levels of Cystatin C and other candidate proteins, providing translational validation of the tumor–brain signaling mechanism.
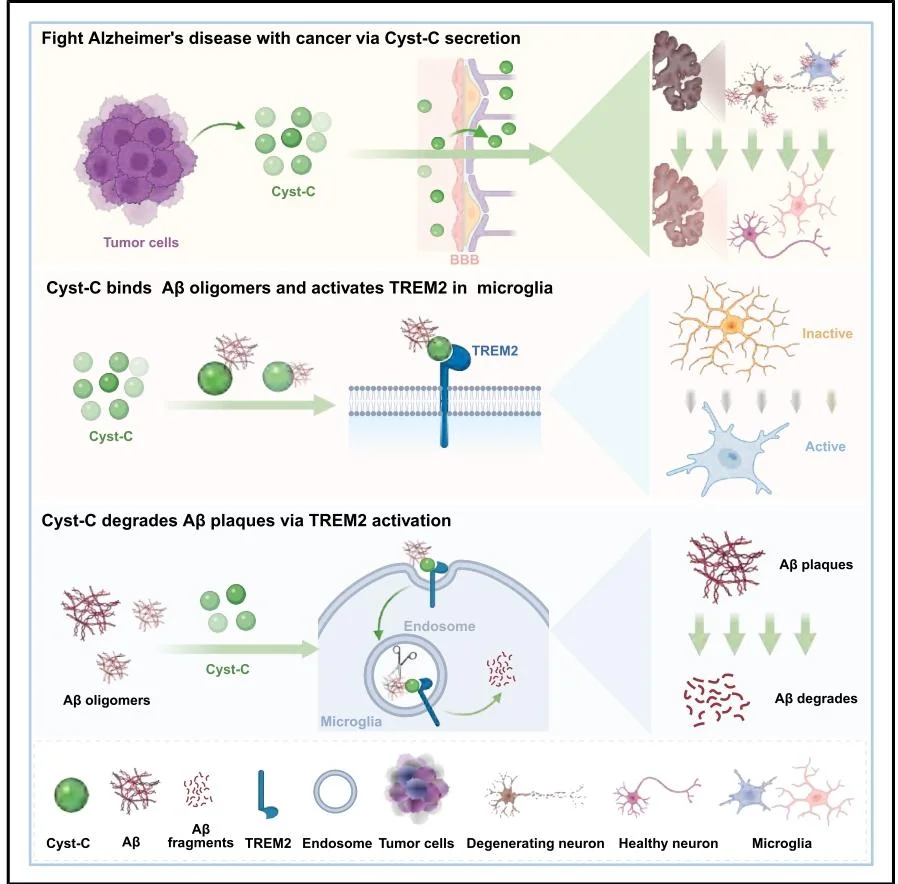
Figure 1. Mechanistic Model: Peripheral Tumor–Derived Cystatin C Counteracts Alzheimer’s Disease Pathology
Research Workflow
- Phenomenon Discovery: In tumor-bearing AD mouse models, multiple peripheral cancers significantly reduced brain Aβ deposition and amyloid plaque burden, while tau misfolding remained unaffected. This finding indicates that peripheral tumors exert a selective inhibitory effect on amyloid pathology in Alzheimer's disease.
- Identification of the Effector Molecule: To exclude the influence of tumor physical burden, the researchers conducted intervention experiments using cancer cell–secreted proteins (CSPs). By integrating transcriptomic and proteomic analyses, they identified Cystatin C (Cyst-C) as the key tumor-derived secreted factor responsible for suppressing amyloid pathology.
- Verification of the Delivery Route: Using ¹²⁵I radiolabeling experiments, the study demonstrated that tumor-derived Cystatin C can penetrate the compromised blood–brain barrier in AD model mice. Importantly, this effect was independent of peripheral immune cell involvement.
- Molecular Mechanism Elucidation: Mechanistic analysis revealed that Cystatin C functions as a ligand for TREM2, directly binding Aβ1–42 oligomers and activating TREM2 signaling on microglia. This activation enhances microglial phagocytosis and degradation of amyloid plaques. Mutations in either TREM2 or Cystatin C completely abolished this effect, confirming the specificity of the signaling pathway.
- Therapeutic Potential Validation: Administration of recombinant human Cystatin C improved neuropathology and cognitive performance in both young and aged AD mouse models in a dose-dependent manner, supporting its therapeutic potential.
Key Findings
1. Peripheral Cancers Selectively Suppress Amyloid Pathology in AD
Three tumor types—Lewis lung carcinoma (LLC), RM1 prostate cancer cell line, and MC38 colon adenocarcinoma cell line significantly reduced soluble and insoluble Aβ1–42 and Aβ1–40 levels, as well as amyloid plaque area in the brains of 5xFAD mouse model mice. However, these tumors did not affect tau phosphorylation in the rTg4510 mouse model, indicating selective modulation of amyloid pathology.
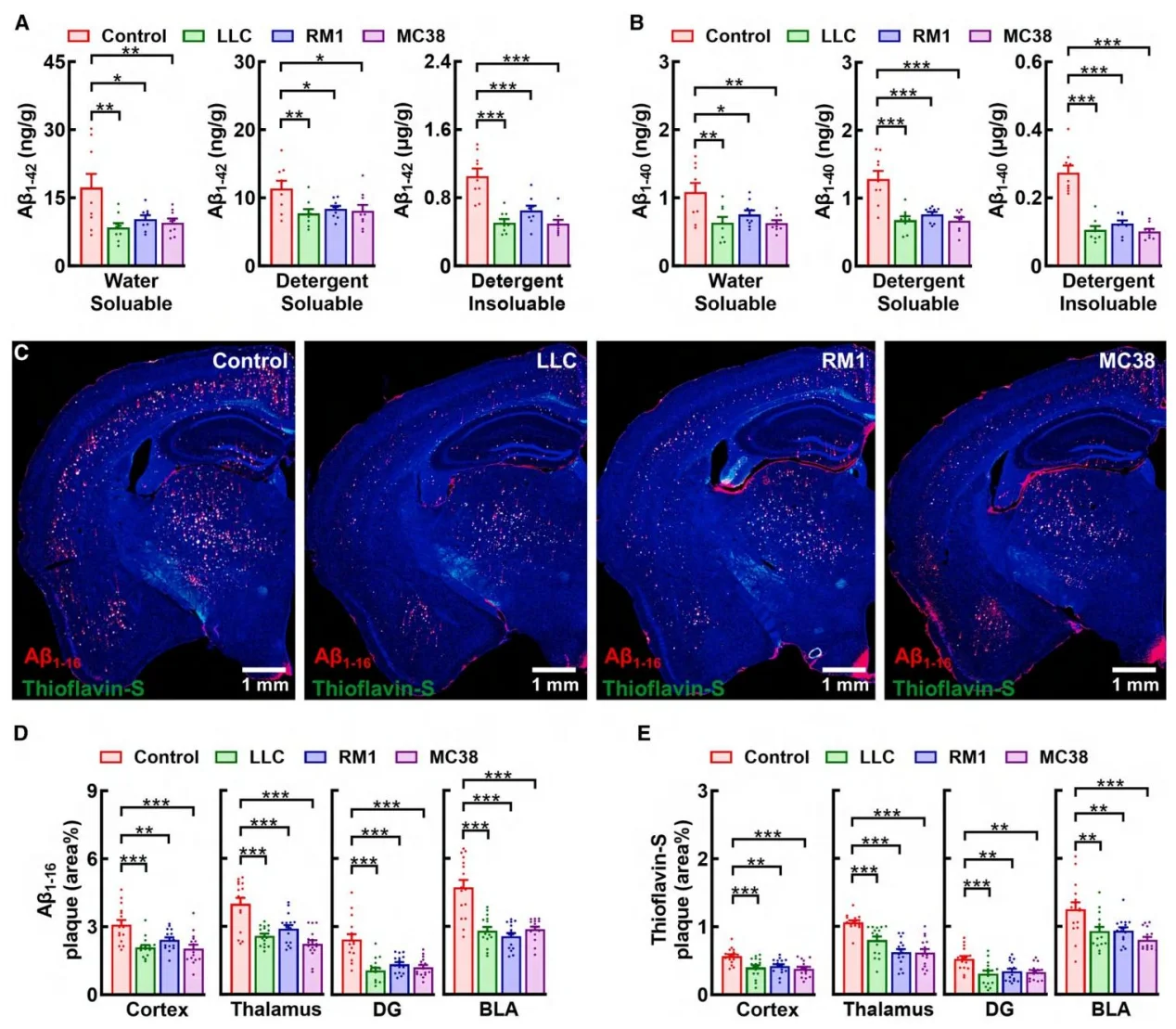
Figure 2. Peripheral cancers alleviate amyloid pathology
2. Cystatin C Is the Key Mediator of the Tumor Protective Effect
Both cancer patients and tumor-bearing mice exhibited significantly elevated plasma levels of Cystatin C. Loss-of-function experiments demonstrated that removal of Cystatin C from CSPs completely abolished their amyloid-suppressing activity. Conversely, supplementation with recombinant human Cystatin C reproduced the protective effect observed in tumor-bearing models. Notably, the Cystatin C L68Q mutant, which cannot bind TREM2, failed to confer protection, confirming the functional importance of the Cystatin C–TREM2 interaction.
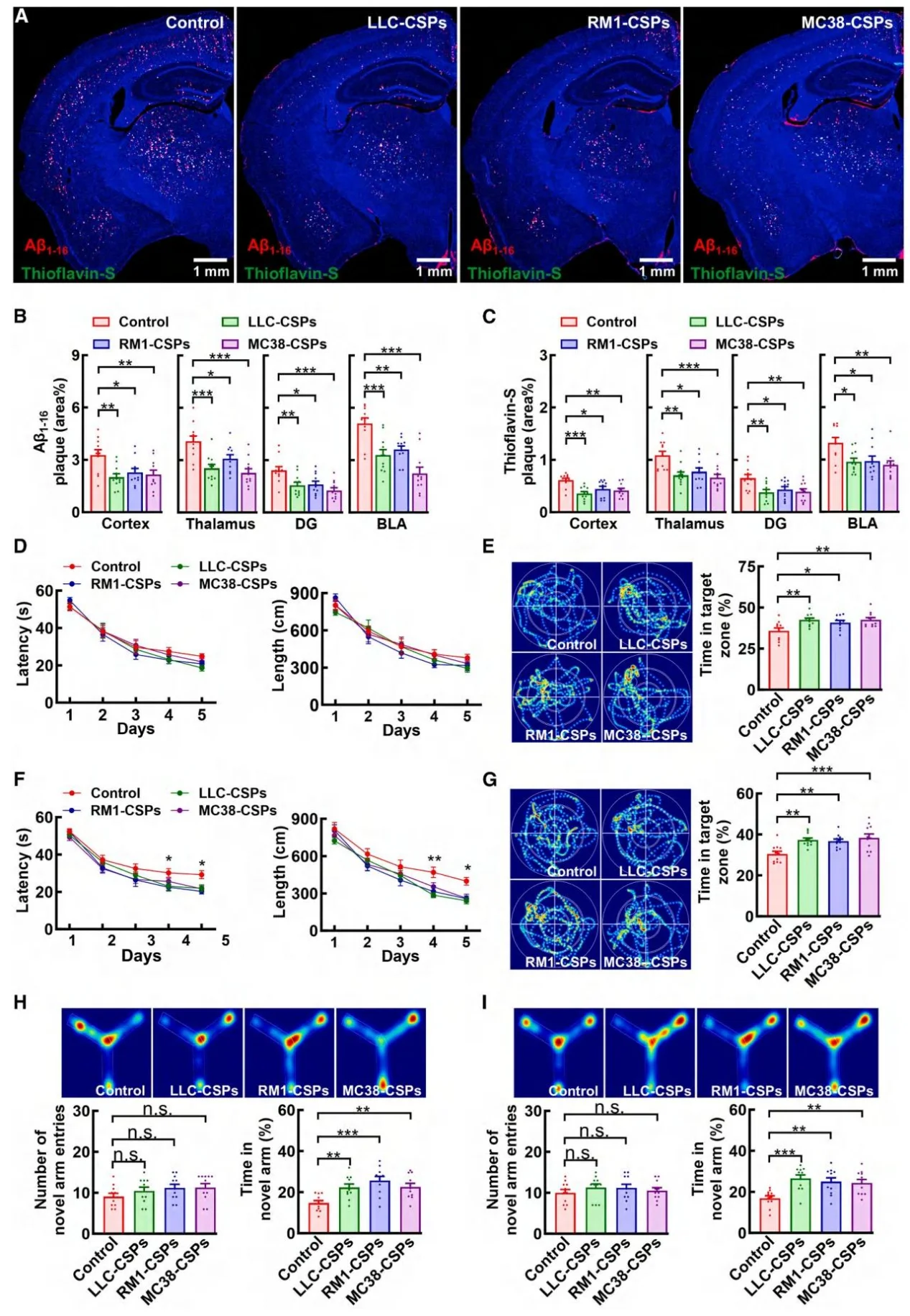
Figure 3. Peripheral cancers suppress amyloid pathology via secreted proteins (CSPs)
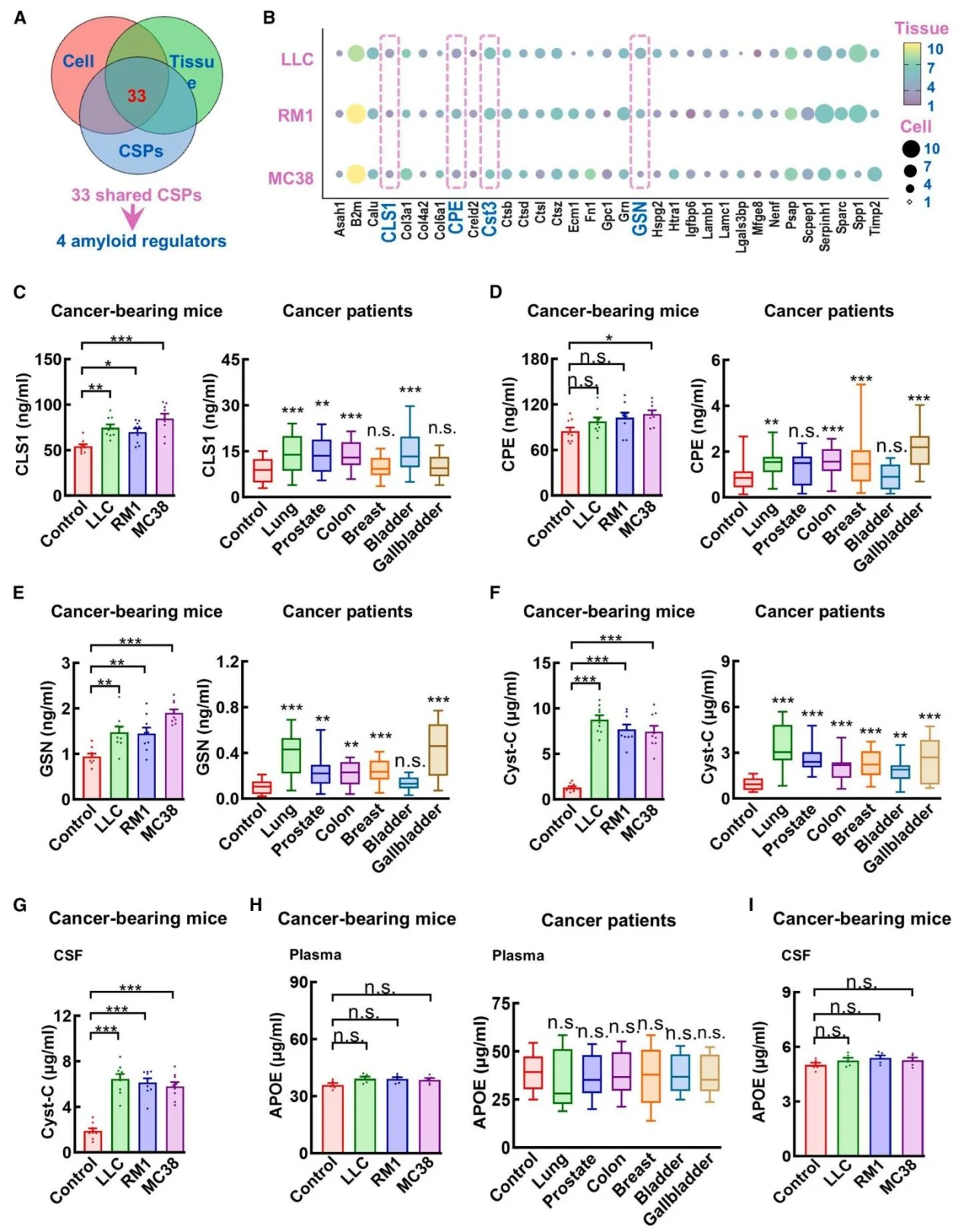
Figure 4. Unbiased proteomic screening of amyloid-suppressing factors within CSPs
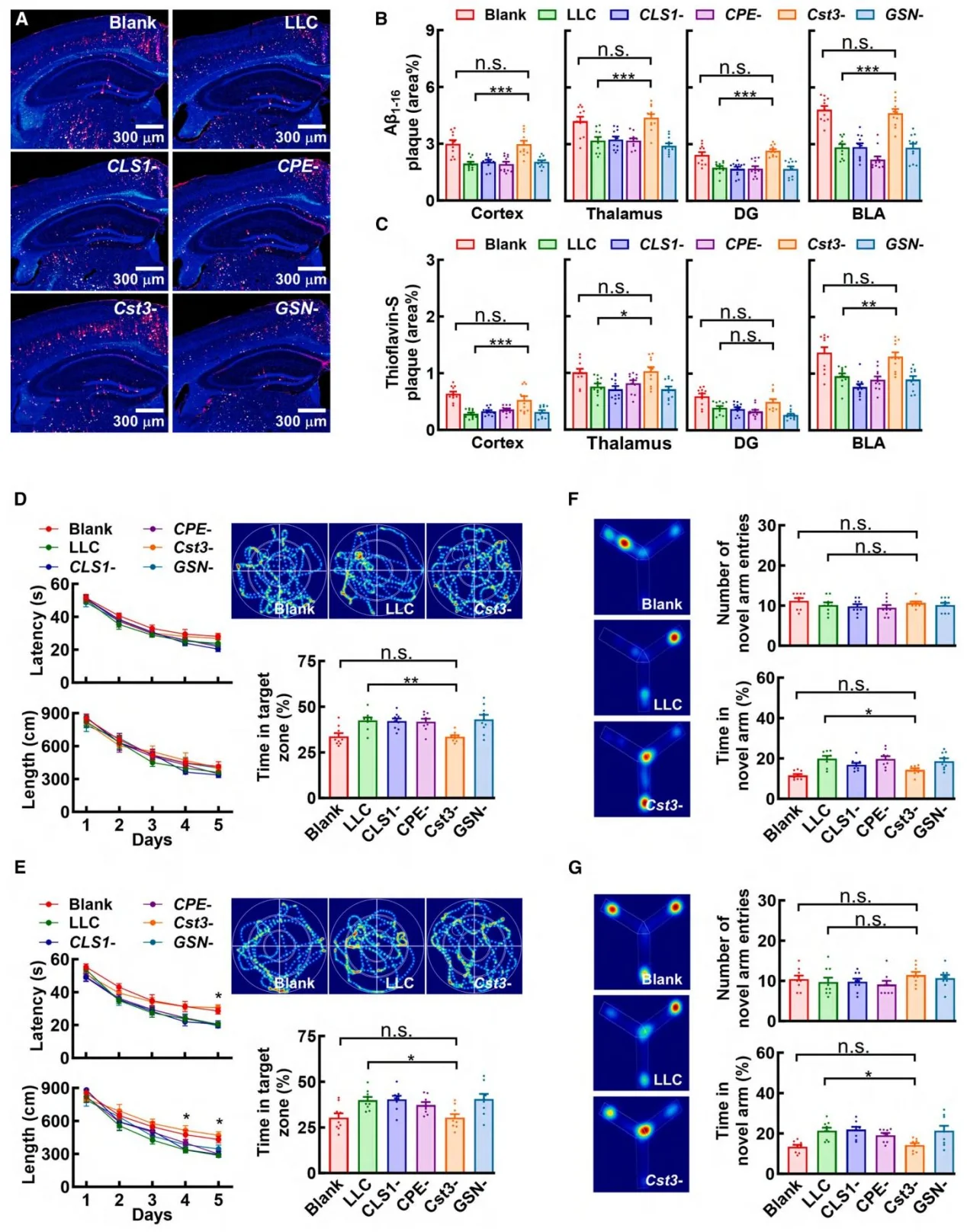
Figure 5. Peripheral cancers inhibit amyloid pathology through secretion of Cystatin C
3. Cystatin C Acts Through the Blood–Brain Barrier
The 13 kDa Cystatin C protein was able to cross the compromised blood–brain barrier in AD model mice, reaching therapeutically relevant concentrations in the brain. In contrast, 67 kDa albumin failed to penetrate the barrier, highlighting the unique delivery advantage of Cystatin C.
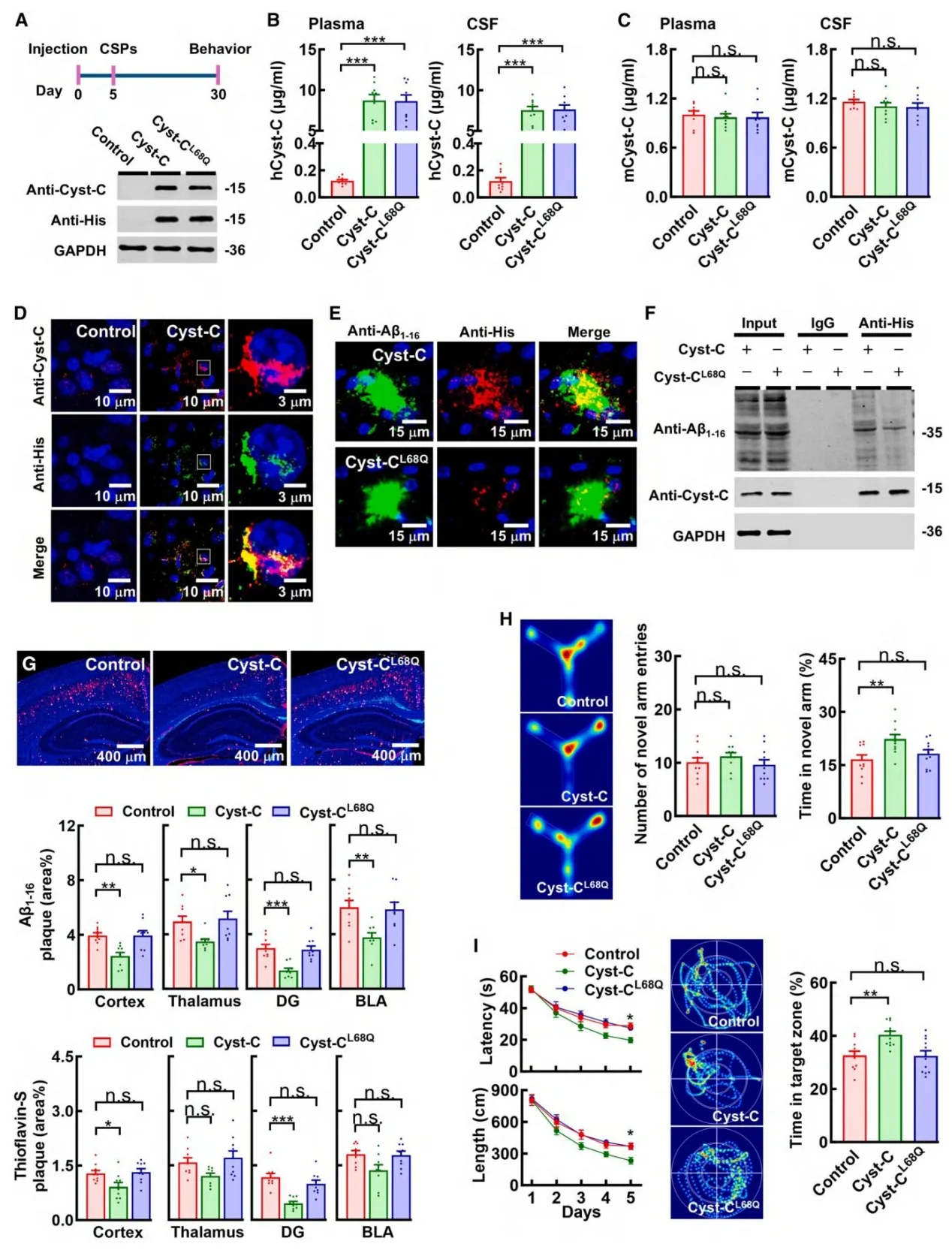
Figure 6. Cancer cell–derived Cystatin C suppresses amyloid pathology
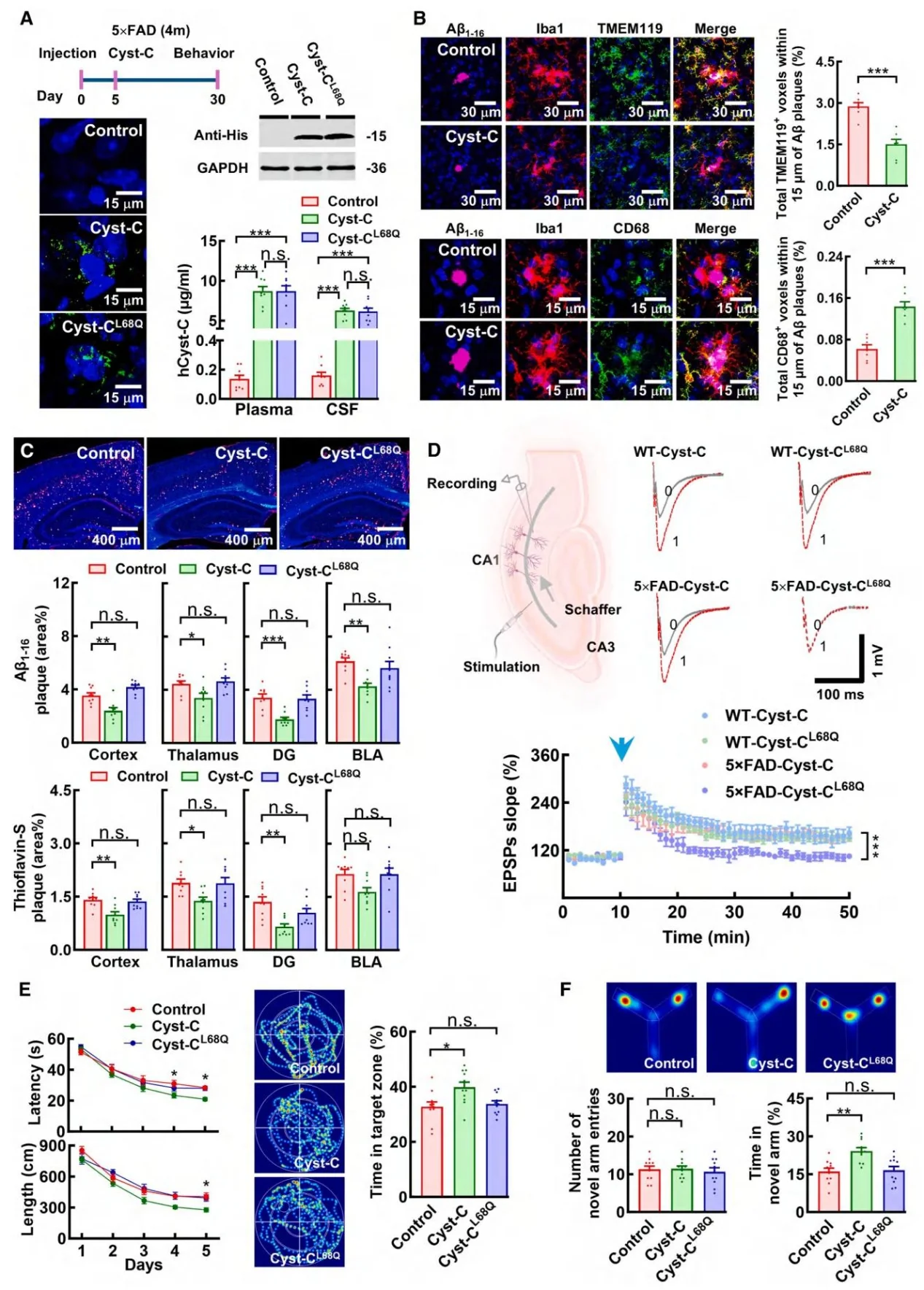
Figure 7. Recombinant human Cystatin C inhibits amyloid pathology
4. The Cystatin C–TREM2 Axis Is the Core Mechanism
Cystatin C directly binds both Aβ1–42 oligomers and the extracellular domain of TREM2. The disease-associated TREM2R47H mutation disrupts this interaction. Activation of TREM2 by Cystatin C triggers the downstream SYK signaling pathway, resulting in: Recruitment of microglia to amyloid plaques, Enhanced microglial phagocytosis, and Accelerated degradation of established plaques. Importantly, microglia-specific knockout of TREM2 completely abolished the therapeutic effects of Cystatin C.
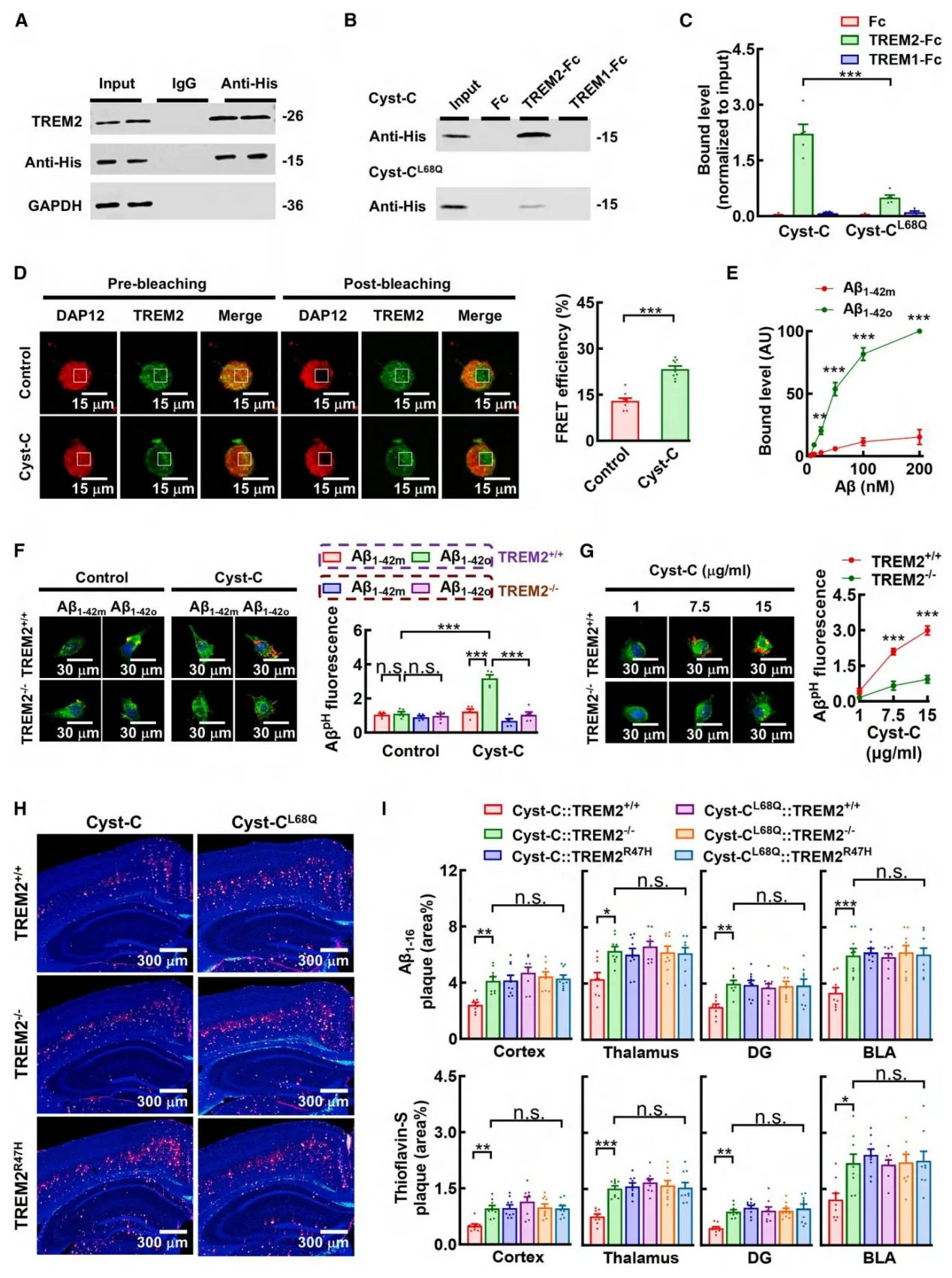
Figure 8. Human Cystatin C promotes degradation of established amyloid plaques via TREM2
5. Cystatin C Shows Strong Translational Potential
Treatment with recombinant human Cystatin C significantly improved cognitive performance across AD mouse models of different ages. It also restored deficits in long-term potentiation (LTP), a key indicator of synaptic plasticity. Notably, these benefits occurred without affecting enzymes involved in Amyloid precursor protein processing, indicating a distinct therapeutic mechanism focused on plaque clearance rather than amyloid production.
Significance and Innovations
- Conceptual Breakthrough: This study demonstrates for the first time that peripheral cancers can actively modulate AD pathology through secreted proteins, establishing a cross-disease regulatory network linking cancer, secreted factors, and neurodegeneration.
- Mechanistic Innovation: The research identifies Cystatin C as a functional agonist of TREM2, revealing a novel pathway: Cystatin C binding to Aβ oligomers → activation of microglial TREM2 → degradation of pre-existing amyloid plaques. This mechanism breaks from traditional AD strategies focused solely on preventing Aβ production.
- New Therapeutic Strategy: The findings propose a plaque-clearance–centered therapeutic strategy for Alzheimer’s disease. Recombinant Cystatin C or its mimetics could potentially serve as precision therapeutics targeting AD pathology, with the added advantage that Cystatin C naturally crosses the blood–brain barrier.
- Clinical Relevance: The study provides a biological explanation for the epidemiological observation of an inverse correlation between cancer and Alzheimer’s disease incidence, offering an experimental foundation for future research into reduced AD risk among cancer patients.
Summary
Through multi-model validation, multi-omics screening, and detailed molecular mechanism analysis, this study demonstrates that peripheral tumors can secrete Cystatin C, which activates the TREM2 signaling pathway to promote degradation of amyloid plaques in Alzheimer's disease mouse models and improve cognitive function. These findings not only reveal a previously unrecognized cross-disease regulatory mechanism between cancer and neurodegeneration, but also challenge conventional therapeutic paradigms in AD research. By identifying the Cystatin C–TREM2 axis as a key driver of amyloid plaque clearance, the study provides a strong experimental foundation for developing novel disease-modifying therapies targeting this pathway. The work therefore holds significant value for both fundamental research and translational medicine.
Ubigene is dedicated to the mission of “Make genome editing easier” Through continuous innovation in technology and service platforms, the company has achieved: 13,000+ successful gene editing projects, 11,000+ engineered cell products, including 8,000+ knockout (KO) cell lines . With proprietary gene-editing technologies, Ubigene has improved editing efficiency by 10–20× compared with conventional methods. To date, the company has supported over 10,000 life science laboratories, pharmaceutical companies, and CRO organizations worldwide with high-quality gene-editing services and products.
The CST3 gene encodes Cystatin C, a key cysteine protease inhibitor involved in regulating inflammation, immune responses, and cellular metabolism. For researchers interested in CST3-related studies, Ubigene offers CST3 knockout cell lines, including widely used cancer models such as: A549 and NCI-H1299. If you require customized gene-editing solutions, Ubigene also provides tailored cell engineering services to support your research projects. For more information about CST3 gene-editing models or custom services, feel free to contact us.
Contact us to learn more>>>Reference
Li X, Tang X, Zeng J, Duan L, Hou Z, Li L, Guo Y, Chai C, Liu J, Wang Y, Zhu LQ, Li H, Zhang T, Wang Y, He A, Lu Y. Peripheral cancer attenuates amyloid pathology in Alzheimer's disease via cystatin-c activation of TREM2. Cell. 2026 Feb 5;189(3):853-871.e31. doi: 10.1016/j.cell.2025.12.020. Epub 2026 Jan 22. PMID: 41576952.
 Promotions
Promotions 








