How to Utilize Gene Editing Technology to Study Point Mutations?

How to Utilize Gene Editing Technology to Study Point Mutations?

In the era of rapidly advancing precision medicine, the study of point mutations has become a crucial direction for unraveling the mechanisms of human diseases and developing targeted therapies. With cutting-edge gene editing technologies such as CRISPR/Cas9, scientists can precisely introduce single nucleotide changes into cellular or animal models, thereby simulating the molecular mechanisms of disease onset and providing powerful tools for basic research and translational medicine.
What is a Point Mutation?
A point mutation refers to a change in a single base within the DNA sequence (including the substitution, insertion, or deletion of a base). Such mutations can occur in protein-coding regions (exons) or non-coding regions (such as promoters, enhancers, introns, and other regulatory regions). Functionally, they can be categorized into loss-of-function and gain-of-function mutations. As illustrated in the figure, coding region point mutations can be divided into synonymous mutations (which do not alter the amino acid) and non-synonymous mutations (which change the amino acid, such as missense or nonsense mutations). Non-coding region point mutations, although not directly altering the protein sequence, may affect gene expression or RNA splicing regulation. Loss-of-function mutations render the gene product inactive or truncated, commonly seen in recessive genetic diseases. In contrast, gain-of-function mutations confer new activity or increased activity to the gene, often associated with dominant diseases or cancers.
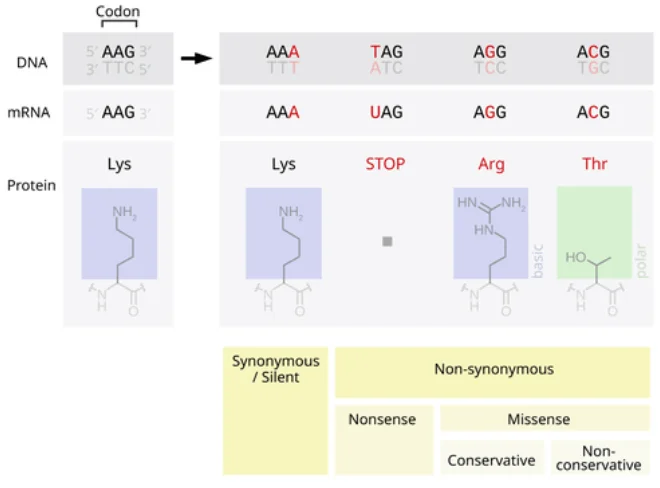
Figure 1. Schematic diagram of the classification of point mutations' impact on proteins. On the left, synonymous mutations do not change the amino acid (Lys→Lys), while on the right are nonsense mutations (generating a stop codon Tag, resulting in a truncated protein) and missense mutations (AA→Arg/Thr, altering the amino acid). The mutated base is indicated in red, with blue and green boxes representing the structural schematic of amino acids.
1. Coding Region Point Mutations: Located in the exons of a gene, these mutations are divided into synonymous mutations (where the protein sequence remains unchanged) and non-synonymous mutations (which alter the amino acid, triggering structural or functional changes).
2. Non-coding Region Point Mutations: Found in regions such as promoters, enhancers, splicing sites, or UTRs, these mutations do not directly alter the protein sequence but may impact gene expression levels, RNA splicing, or the function of regulatory elements.
3. Loss-of-Function Mutations: The gene product becomes inactive or truncated, such as when a nonsense codon is generated, leading to the loss of protein function. These mutations are often recessive (requiring mutations in both alleles to cause disease).
4. Gain-of-Function Mutations: The mutation endows the gene with new functionality or over-activation, commonly seen in dominant diseases or cancer genes. For instance, mutations in oncogenes can lead to continuous cell proliferation.
The Principle of CRISPR/Cas9 in Creating Point Mutation Models
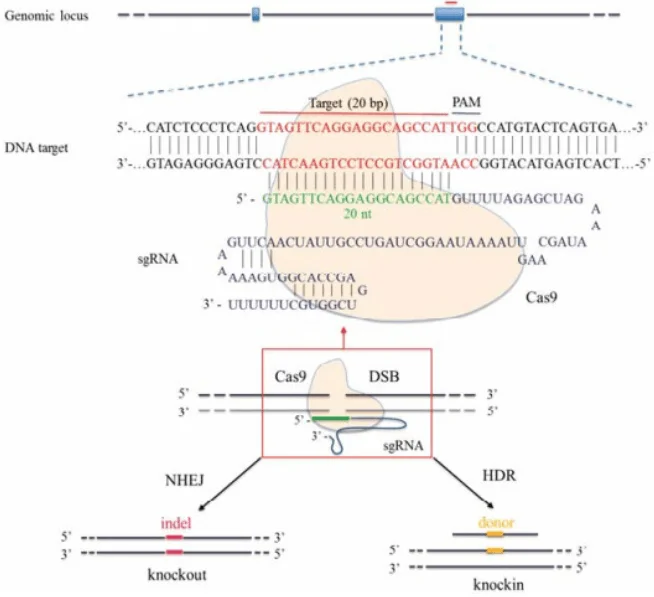
Figure 2: Schematic diagram of CRISPR/Cas9-mediated gene editing. The Cas9 protein (yellow) recognizes the target sequence (blue) under the guidance of sgRNA and cleaves the DNA, causing a double-strand break (DSB). The cell initiates repair mechanisms; non-homologous end joining (NHEJ) often introduces small insertions/deletions (indels) leading to gene knockout (left), while homologous recombination (HDR) utilizes the provided donor DNA (yellow fragment) to precisely repair and insert specific mutations (right).
The CRISPR/Cas9 system can induce double-strand breaks (DSBs) at specific genomic sites and leverage the cell's own repair mechanisms to modify the genome. As shown in the figure, after Cas9 cuts the DNA under the guidance of a single-guide RNA (sgRNA), if no donor template is provided, the cell's NHEJ repair often results in random insertions/deletions (indels), which can cause frame-shifts or premature termination, effectively knocking out the gene. However, if a homologous recombination repair template containing the desired mutation (such as a double-stranded DNA or single-stranded oligonucleotide with the mutation site) is introduced simultaneously in the experiment, the cell can precisely "repair" the mutation into the genome via HDR, thereby achieving point mutations or gene knock-ins.
Based on this principle, we can create cell lines or animal models carrying specific point mutations: by designing a pair of sgRNA/Cas9 to target the desired site and providing donor DNA containing the target mutation, the cell's HDR repair mechanism will incorporate the mutation into the genome. After obtaining the edited mixed cells, pure or heterozygous mutant clones are typically isolated through single-cell cloning or other screening methods. In polyploid cells or animal embryos, if the editing efficiency is high, both alleles may be edited simultaneously, resulting in a homozygous mutation; if only one allele is edited, a heterozygous mutant model is obtained. For dominant genetic diseases, constructing a heterozygous mutant is often sufficient to simulate the patient's genotype; for recessive genetic diseases, a homozygous model with mutations in both alleles is required.
Case Studies of Disease Models
· Centronuclear Myopathy (CNM, Heterozygous Mutation)
Autosomal dominant CNM is commonly caused by a single point mutation in the DNM2 gene, such as R465W. Rabbi et al. designed an sgRNA/Cas9 system that specifically recognizes the mutant allele in patient-derived myoblasts and mouse models, successfully targeting and knocking out the mutant DNM2 allele. Post-editing, the abnormal cellular membrane transport and autophagy functions were restored. This demonstrates that CRISPR/Cas9 can perform allele-specific editing of dominant mutant alleles, thereby reversing the disease cell phenotype and providing possibilities for the treatment and study of dominant point mutation diseases.
· Alzheimer's Disease (AD, Heterozygous Mutation)
Familial early-onset AD is often triggered by point mutations in the APP or PSEN genes, such as the Swedish mutation (APP KM670/671NL) or the PSEN1 M146L mutation. Researchers utilized CRISPR/Cas9 to design specific gRNAs that only disrupt the mutant allele. In patient fibroblasts carrying the Swedish mutation, knocking out the mutant APP allele reduced the level of Aβ peptide in the culture supernatant by approximately [X]% (the exact percentage should be specified). Similarly, after gene editing in cells carrying the PSEN1 M146L heterozygous mutation, the elevated Aβ42/40 ratio significantly decreased and partially returned to normal. These results indicate that CRISPR editing targeting AD-related point mutations can alleviate pathogenic phenotypes, offering new insights for Alzheimer's disease mechanism research and therapeutic exploration.
· Tay-Sachs Disease (TSD, Homozygous Mutation)
TSD is an autosomal recessive neurodegenerative disease, with 80% of patients carrying a four-base (TATC) insertion mutation in exon 11 of the HEXA gene. Knocking out the HEXA gene in mice failed to reproduce the typical pathology. Therefore, Qian et al. attempted to use gene editing technology to construct a TSD model. They ultimately employed the next-generation prime editing technology to insert the TATC mutation into the rabbit HEXA gene, successfully obtaining a rabbit model carrying this insertion mutation and observing phenotypes similar to human TSD, such as muscle weakness and ataxia. This example illustrates that by precisely editing human pathogenic point mutations into animal models, the corresponding genetic disease phenotypes can be reproduced in vivo, providing tools for in-depth disease mechanism studies.
· Cystic Fibrosis (CF, Homozygous Mutation)
CF is a lethal genetic disease caused by recessive mutations in the CFTR gene, with point mutations such as ΔF508 causing loss of CFTR protein function. Fan et al. applied CRISPR/Cas9 and somatic cell nuclear transfer technology in sheep to knock out the sheep CFTR gene. They successfully obtained CFTR^-/- and CFTR^+/- sheep, with CFTR^-/- sheep exhibiting pancreatic fibrosis, intestinal obstruction, and absence of the vas deferens, which are highly consistent with the pathological manifestations of human CF. This large animal model demonstrates that CFTR gene knockout is sufficient to induce typical CF phenotypes and validates the potential of this model for testing new drugs and gene therapies.
The Significance of Point Mutation Disease Models
Utilizing CRISPR/Cas9 technology to construct disease models carrying specific point mutations helps accurately simulate human pathogenic genotypes, providing a robust platform for studying disease mechanisms and validating therapeutic strategies. As domestic scholars have pointed out, CRISPR/Cas9 has enabled targeted gene editing, making the construction of animal or cell line models more convenient and significantly accelerating drug target screening, validation, and new drug development. For instance, the aforementioned CF sheep model and TSD rabbit model not only reproduce the key pathology of human diseases but can also be used to evaluate the efficacy of gene therapies or drugs targeting mutations in vivo. Modern new drug development increasingly relies on precise disease models: through point mutation models, we can identify key signaling pathways in disease mechanism research and test the efficacy of candidate drugs on specific mutations during drug screening, thereby laying the foundation for the development of personalized medicine and gene therapy strategies.
The Future Potential of New Gene Editing Platforms
With continuous technological iteration, the next generation of gene editing tools is bringing more possibilities to point mutation research. The EZ-editor™ platform and EZ-HRex™ launched by Ubigene Biosciences achieve gene editing efficiencies 10 to 20 times higher than traditional CRISPR/Cas9, enabling efficient acquisition of gene knockouts, point mutations, and gene knock-ins. In addition, base editing and prime editing technologies can directly achieve single base substitutions without causing double-strand breaks in DNA, offering new approaches for correcting or simulating point mutations. In the future, these highly efficient and precise gene editing platforms will greatly simplify the process of constructing disease models, allowing researchers to more rapidly generate cell lines and animal models with various complex mutation backgrounds. This will not only accelerate basic scientific research but also expand new avenues for gene therapy and personalized drug development.

EZ-HRex™ New Technique for point mutation
In summary, gene editing technologies, especially CRISPR/Cas9 and its derivative platforms, have provided powerful tools for the study of point mutations. From exploring mutation mechanisms to constructing models and screening drugs, scientists are fully utilizing these technologies to advance life science research and look forward to the near future when these emerging platforms can more efficiently crack genetic diseases, creating more possibilities for the development of precision medicine and innovative therapies.
References
[1] Rabai A, Reisser L, Reina-San-Martin B, Mamchaoui K, Cowling BS, Nicot AS, Laporte J. Allele-Specific CRISPR/Cas9 Correction of a Heterozygous DNM2 Mutation Rescues Centronuclear Myopathy Cell Phenotypes. Mol Ther Nucleic Acids. 2019 Jun 7;16:246-256. doi: 10.1016/j.omtn.2019.02.019. Epub 2019 Feb 27. PMID: 30925452; PMCID: PMC6439232.
[2] Konstantinidis E, Molisak A, Perrin F, Streubel-Gallasch L, Fayad S, Kim DY, Petri K, Aryee MJ, Aguilar X, György B, Giedraitis V, Joung JK, Pattanayak V, Essand M, Erlandsson A, Berezovska O, Ingelsson M. CRISPR-Cas9 treatment partially restores amyloid-β 42/40 in human fibroblasts with the Alzheimer's disease PSEN1 M146L mutation. Mol Ther Nucleic Acids. 2022 Mar 28;28:450-461. doi: 10.1016/j.omtn.2022.03.022. PMID: 35505961; PMCID: PMC9043867.
[3] György B, Lööv C, Zaborowski MP, Takeda S, Kleinstiver BP, Commins C, Kastanenka K, Mu D, Volak A, Giedraitis V, Lannfelt L, Maguire CA, Joung JK, Hyman BT, Breakefield XO, Ingelsson M. CRISPR/Cas9 Mediated Disruption of the Swedish APP Allele as a Therapeutic Approach for Early-Onset Alzheimer's Disease. Mol Ther Nucleic Acids. 2018 Jun 1;11:429-440. doi: 10.1016/j.omtn.2018.03.007. Epub 2018 Mar 16. PMID: 29858078; PMCID: PMC5992788.
[4] Fan, Z., et al. "A sheep model of cystic fibrosis generated by CRISPR/Cas9 disruption of the CFTR gene. JCI Insight 3: e123529." 2018
 Promotions
Promotions 








