How to study protein kinase by CRISPR technology?

Protein kinases (PK) and their opponents, phosphatases, have been crucial to many scientists. Approximately 2% of the human genome encodes for PKs known as human “kinome”, consisting of 518 protein kinases and their variants. Protein kinases are key regulators of cell function. They mediate most of the signal transduction in eukaryotic cells and coordinate the activity of multiple cellular processes including metabolism, transcription, cell cycle progression, cell movement, differentiation, and apoptosis. They direct the activity, localization, and overall function of many proteins by modifying protein activity, though adding phosphate groups to their substrate.
Mutations and dysregulation of protein kinases play causal roles in human disease, particularly cancer. Their involvement in multiple aspects of cell biology opens the possibility of developing agonists and antagonists for therapeutic use. A growing interest in developing orally active protein kinase inhibitors has led to the approval of several inhibitors for clinical use.
Protein kinases are among the largest and most well-studied gene families. Protein phosphorylation plays an essential role in intercellular communication in eukaryotic organisms by mediating signal transduction during development, transcription, immune response, metabolism, apoptosis, and cell differentiation. Aberrant regulation of kinases plays a causal role in many diseases, and the study of these proteins and their functions will contribute to the discovery and development of new therapeutics. kinome allows an accurate study of the kinase‐tumor dependent phenotype and, therefore, has the potential to reveal potential therapeutic targets.
The human protein kinome presents one of the largest protein families that orchestrate functional processes in complex cellular networks, and when perturbed, can cause various cancers. The abundance and diversity of genetic, structural, and biochemical data underlie the complexity of mechanisms by which targeted and personalized drugs can combat mutational profiles in protein kinases. To date, PKs are validated and widely accepted targets for drug discovery, more than 40 drugs have been FDA-approved and are on the market. The majority of compounds address tyrosine kinases, with their main therapeutical applications being cancer and inflammation. Coupled with the evolution of system biology approaches, genomic and proteomic technologies are rapidly identifying and characterizing novel resistance mechanisms to inform the rationale design of personalized kinase drugs.
Application:
1. Network: Studying the kinome as a network of kinases or kinomics
2. Disease development: Study on Kinases in disease developments (cancer research kinases, cardiovascular disease research kinases, kinases for nervous system research i.e Alzheimer research kinases, Parkinson research kinases, multiple sclerosis kinases, etc, metabolic disorder research, and respiratory research).
3. Drug discovery: Study of identifying kinase substrates, kinase drug response, drug discovery, mutant kinases & downstream effects, disease mechanism. Drug design by unraveling complex relationships between the robustness of targeted kinase genes and binding specificity of targeted kinase drugs. Develop novel strategies for rationally tailored and robust personalized drug therapies.
4. Disease model: Kinase disease models (Cancer, cardiovascular disease, Alzheimer, research kinases, Parkinson, multiple sclerosis, metabolic, and respiratory, etc).
Case study:
Serotonin receptor 5A is a potential target for anticancer drug development
Breast tumor-initiating cells (BTIC) are stem-like cells that initiate and sustain tumor growth and drive disease recurrence. Identifying therapies targeting BTIC has been hindered due primarily to their scarcity in tumors. Some small molecules can affect BTIC survival. In this study, the researcher demonstrates that exposure of human breast tumor cells to several structurally unrelated selective antagonists of 5-HT5A reduced BTIC frequency and that this effect was phenocopied by a CRISPR-Cas9-mediated knockout of HTR5A. They used a phosphoproteomic approach to establish that exposure of human breast tumor cells to SB-699551 disrupts signaling via the Gα i/o a coupled pathway and the PI3K/AKT/mTOR axis, consistent with antagonism of 5-HT5A. They showed that SB-699551 reduced human breast tumor xenograft growth rate and functioned in concert with docetaxel chemotherapy to shrink the xenografts. Collectively they provide genetic, pharmacological, and phosphoproteomic evidence that 5-HT5A is the likely target of SB-699551 and that selective 5-HT5A antagonists might be developed into a novel class of anti-cancer agents that can be combined with cytotoxic therapies to shrink established breast tumor xenografts.
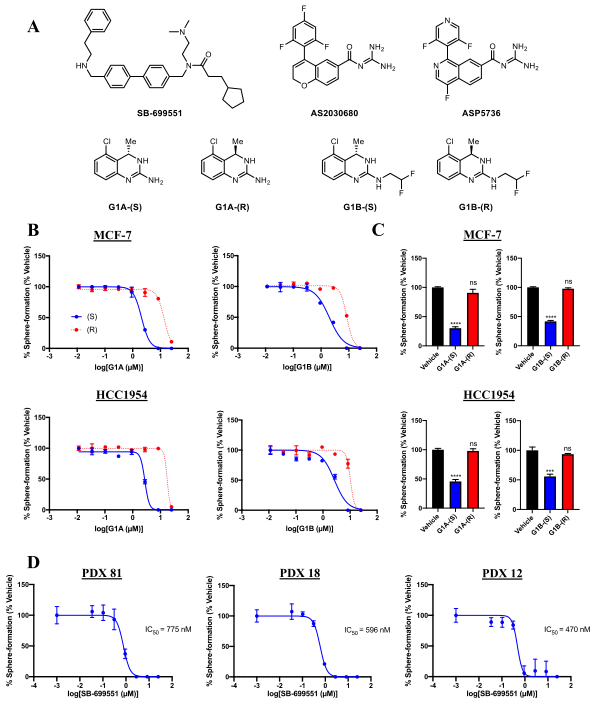
Figure-1: Selective antagonists of 5-HT5A inhibit tumorsphere formation by human breast tumor cells.
The researcher used structurally unrelated 5-HT5A selective antagonists that reduce the frequency of tumorsphere initiating cells in breast cancer cell lines and those derived from human patient-derived tumors (Figure1).
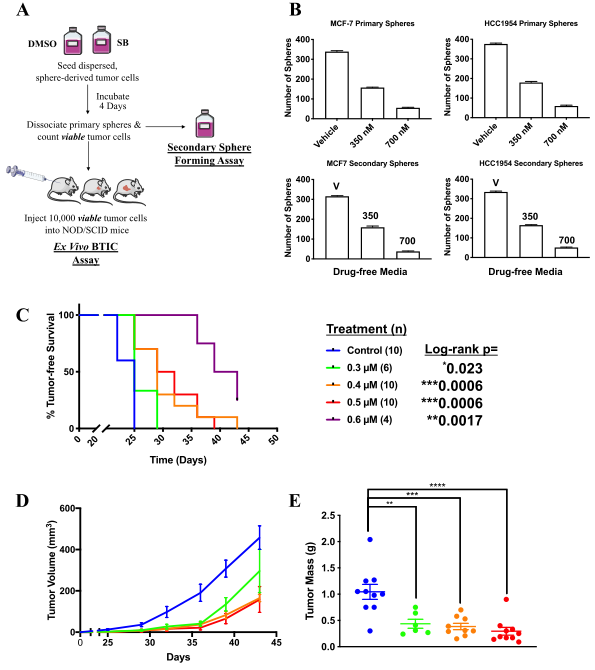
Figure 2: SB-699551 affects tumorsphere formation by an irreversible mechanism and targets BTIC.
The researcher observed that SB-699551 inhibits tumorsphere formation by an irreversible mechanism and BTIC frequency per se was affected by exposure to SB-699551, we assessed its capacity to affect tumor initiation using ex vivo assay. They found that xenograft formation significantly delayed in cohorts engrafted with tumor cells exposed to SB-699551 in a dose-dependent fashion as well as xenografts arising from SB-699551-treated tumor cells exhibited a reduced growth rate and mass (Fig 2).
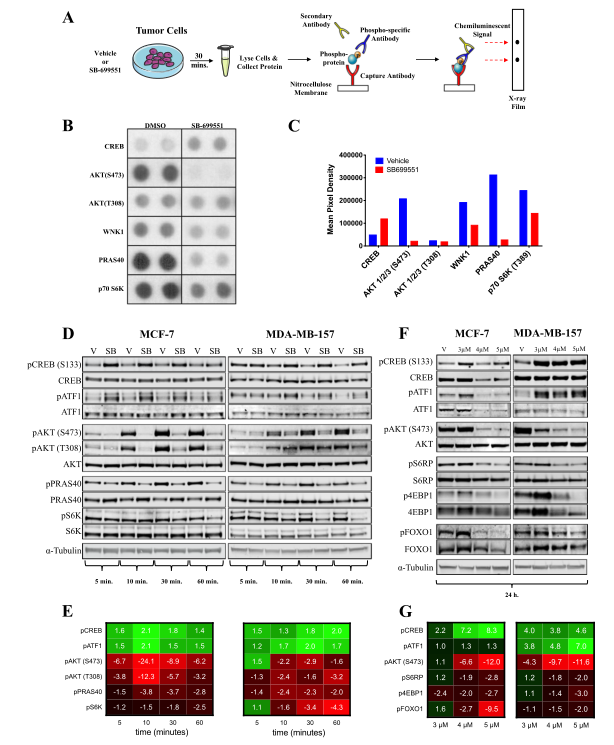
Fig 3: SB-699551 signals via canonical Gα i/o coupling and through the PI3K/AKT/mTOR pathway.
To determine the effect of SB-699551 on BTIC survival, cells were treated with the compound and identify the signaling pathways that might be affected in breast tumor cell lines after treatment with the compound. They used the PPA, which measures the phosphorylation status of 43 intracellular proteins using phospho-specific antibodies in a sandwich ELISA format. The phosphoproteins whose abundance was most affected by treatment with SB-699551 was shown in Fig. 3b-c. Interestingly, the decreased phosphorylation of pAKT, pCREB, and pATF1 observed previously was maintained after 24h of SB-699551 treatment (Fig.3f-g).
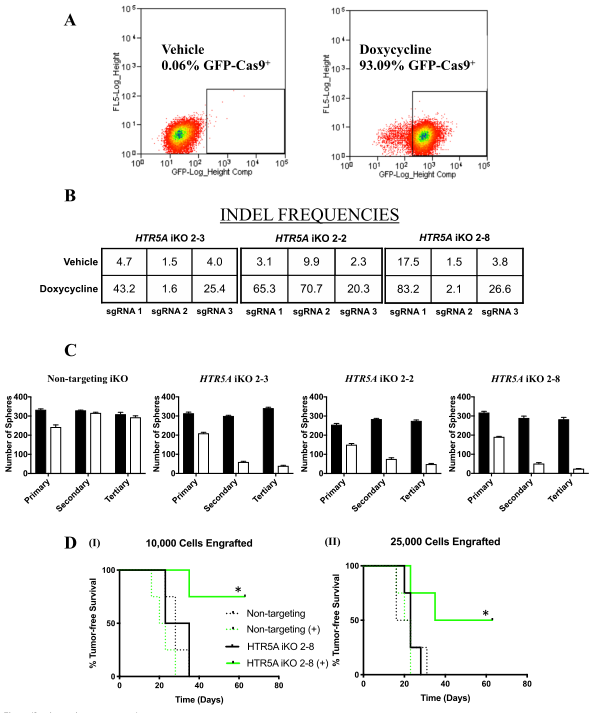
Figure 4: Inducible knockout of HTR5A affects tumorsphere formation and targets BTIC
To investigate the genetic loss of HTR5A phenocopy and their effects in functional assays, we used the CRISPR-Cas9 gene-editing technology to conditionally mutate HTR5A in MCF-7 breast tumor cells. MCF-7 cell lines (2-2, 2-3, and 2-8) were generated by dox-induced Cas9 mediated insertions/deletions (INDELS) in HTR5A (inducible knockout; iKO), as established by next-generation sequencing of genomic DNA (Fig4b). To ensure that BTIC frequency per se was affected by the loss of 5-HT5A activity, they performed ex vivo assays with the MCF-7 NT iKO and HTR5A iKO 2–8 cell line.
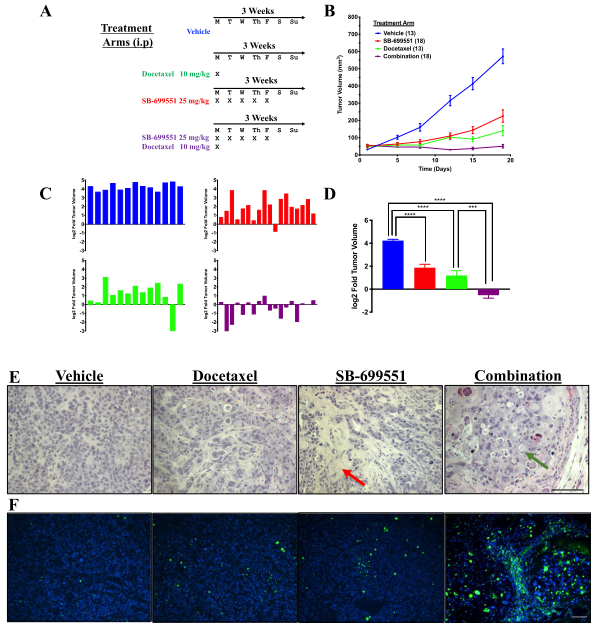
Figure 5: Treatment with SB-699551 inhibits the growth of human breast tumor xenografts in vivo
SB-699551 reduces the growth rate of human breast tumor xenografts in NOD/SCID mice when administered alone and shrinks the xenografts in combination with docetaxel. Histological examination and TUNEL assays revealed an increase in the frequency of apoptotic tumor cells in the xenografts of mice treated with a combination of both agents. The effect of each compound on tumor growth is consistent with SB-699551 and docetaxel targeting BTIC and their non-tumorigenic progeny respectively (Figure 5).
In summary, SB-699551 reduced 5-HT5A activity by compromising its capacity to signal to downstream effectors known to be dysregulated in breast and other cancers. Thus, 5-HT5A is a suitable molecular target for anticancer drug development.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!
Reference:
Gwynne, William D., et al. "Antagonists of the serotonin receptor 5A target human breast tumor initiating cells." BMC cancer 20.1 (2020): 1-17.

 Subscribe Us
Subscribe Us Gene Editing Services
Gene Editing Services
 EZ-editor™
EZ-editor™ Red Cotton Gene knockout Project
Red Cotton Gene knockout Project





