Riboflavin Metabolism Shapes FSP1-Driven Ferroptosis Resistance


Introduction
Ferroptosis suppressor protein 1 (FSP1) serves as a secondary line of defense against cellular ferroptosis; however, upstream regulators governing FSP1 remain insufficiently characterized. Recently, a study published in Nature Cell Biology by José Pedro Friedmann Angeli’s group at the University of Würzburg demonstrated that riboflavin (vitamin B2) metabolism acts as a core determinant of FSP1 protein stability and ferroptosis sensitivity. Within this metabolic cascade, riboflavin kinase (RFK) and FAD synthetase (FADS) represent critical nodes required to sustain FSP1 protein stability and its antioxidant capacity. Furthermore, the study revealed that roseoflavin, a riboflavin antimetabolite, can be metabolically integrated into FSP1 by host cells. Although roseoflavin incorporation successfully preserves FSP1 protein stability, it renders the enzyme catalytically inactive, thereby sensitizing cancer cells to ferroptosis. This discovery offers a promising therapeutic rationale for targeting the FSP1-Coenzyme Q10 (CoQ10) antioxidant axis in oncology.

Research Background
Cells primarily rely on glutathione peroxidase 4 (GPX4) and ferroptosis suppressor protein 1 (FSP1) to suppress ferroptosis induction. FSP1 utilizes NAD(P)H as an electron donor to halt the radical chain propagation of lipid peroxidation. While the downstream execution mechanism of FSP1 is well established, the actionable upstream factors modulating its functional status remain largely uncharacterized.
Research Objectives
- To systematically screen for upstream functional regulators of FSP1.
- To validate RFK and FAD as direct modulators of FSP1 and elucidate their underlying biochemical mechanisms.
- To determine whether riboflavin bioavailability serves as a pivotal baseline metric for ferroptosis sensitivity.
- To clarify the sensitizing mechanism of the riboflavin antimetabolite roseoflavin and evaluate its translational potential in oncology.
Research Method
- Cellular Models: Overexpression (OE) cell lines for FSP1, FADS, and SLC52A2 (established in HT1080 and A375 backgrounds); Knockout (KO) cell lines including GPX4KO (HT1080) , FSP1KO (A375, MDA-MB-231) , RFKKO (A375) , FLAD1KO (A375) , SLC52A2KO (HT1080) , and the engineered GPX4 KO/FSP1OE HT1080 CRISPR library screening model.
- CRISPR Screening: A customized FSP1 target modifier library containing 15,062 unique sgRNAs was engineered. Transduction was performed on GPX4 KO/ FSP1 OE HT1080 cells, followed by Next-Generation Sequencing (NGS) to isolate positive hits.
- Mechanistic Validation: Western blotting (WB) was deployed to assess FSP1 protein destabilization following RFK ablation. Global proteomic profiling was utilized to uncover structural polypeptide backbone instability within FSP1 under FAD-depleted conditions.
Research Results
1. CRISPR Screen Identifies RFK as an Essential FSP1 Regulator
SgRNAs targeting RFK exhibited a pronounced dropouts (depletion) upon the withdrawal of Lip-1, qualifying it as a primary candidate gene. Immunoblotting confirmed that genetic ablation of RFK led to a striking decrease in baseline FSP1 protein levels. Consequently, RFKKO cells displayed marked hypersensitivity to the covalent GPX4 inhibitors RSL3 and ML210. This hyper-sensitization profile was fully rescued by the lipophilic antioxidant Lip-1, confirming that the cell death phenotype is driven exclusively by the ferroptotic pathway.
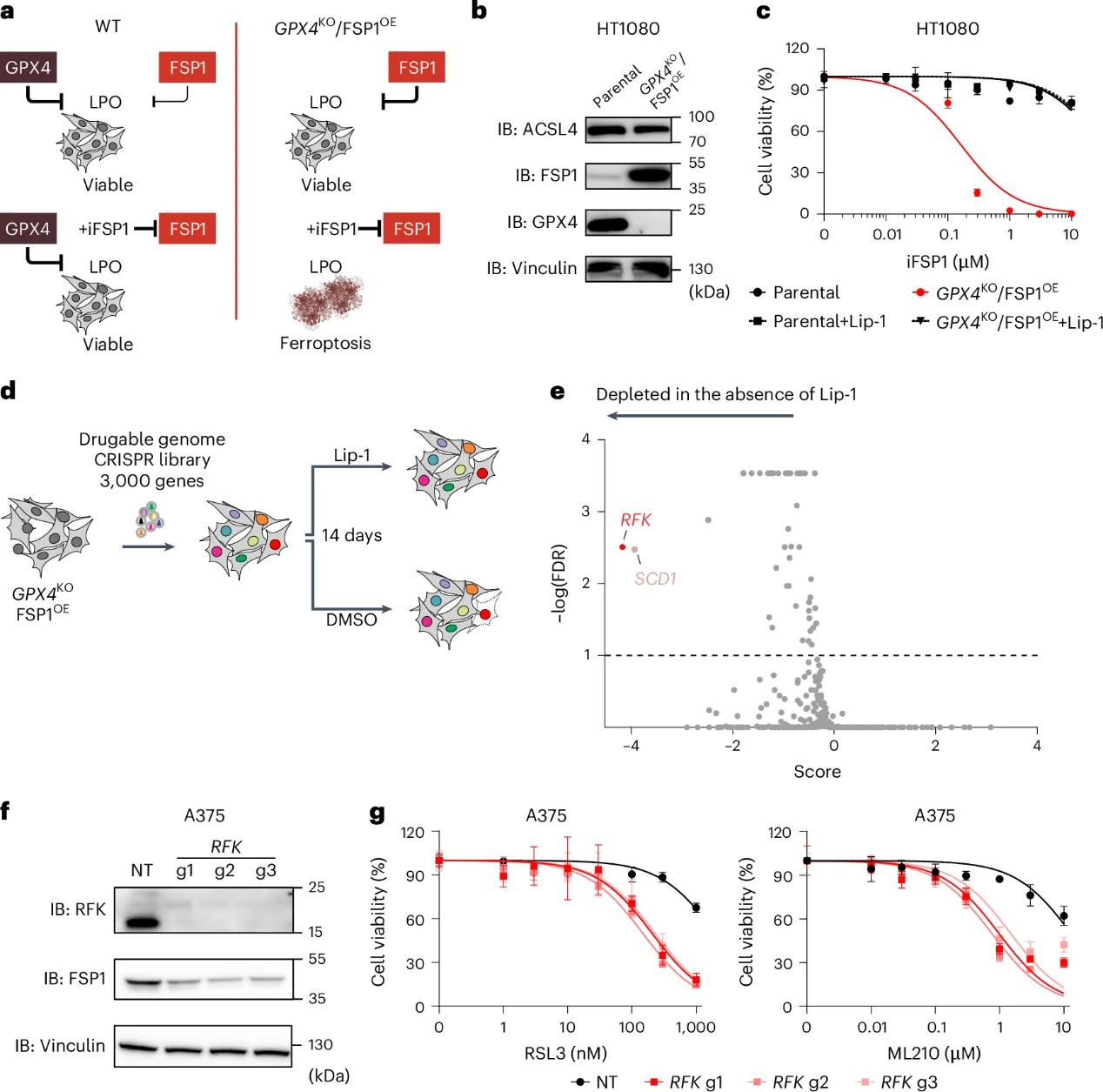
Fig. 1: Identification of factors supporting FSP1 function
2. FAD Deficiency Abrogates FSP1 Function and Augments Ferroptosis Sensitivity
Depletion of intracellular FAD heightened cellular sensitivity to GPX4 inhibition and accelerated lipid peroxidation. Proteomic analysis showed a severe loss of flavoproteins, notably FSP1 and NQO1, establishing that FAD deficiency compromises FSP1 structural stability and acts as a baseline amplifier for ferroptotic stress.
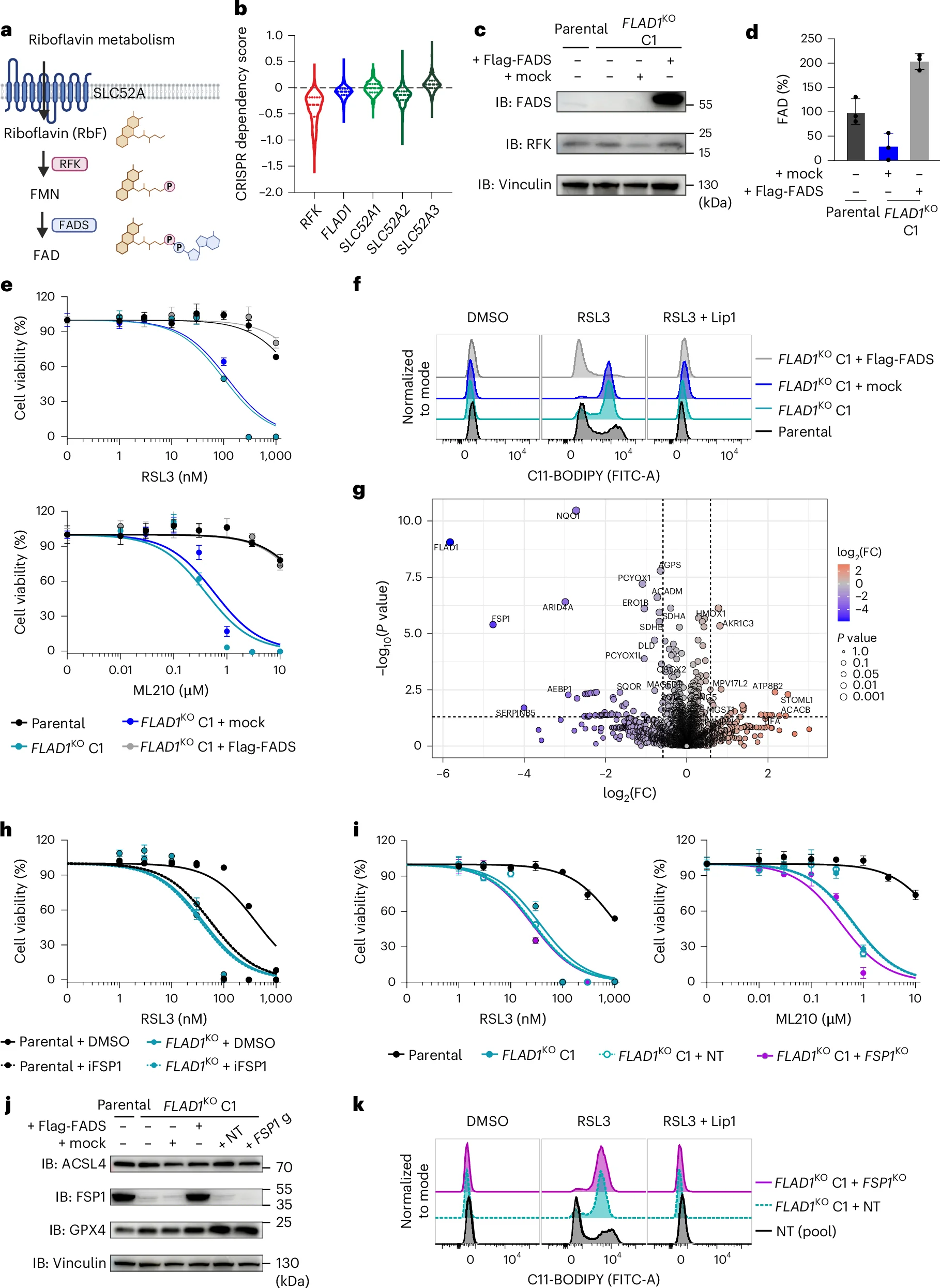
Fig. 2: FAD deficiency disrupts FSP1 function and promotes ferroptosis susceptibility
3. Riboflavin Availability is a Core Determinant of Ferroptosis Resistance
As the direct metabolic precursor to FAD, riboflavin restriction triggered a severe down-regulation of FSP1 expression, yielding a profound vulnerability to lipid peroxidation. Importantly, this riboflavin-depletion-mediated sensitization was entirely blunted in FSP1KO cell lines, demonstrating that riboflavin exerts its anti-ferroptotic protection via an FSP1-dependent axis.
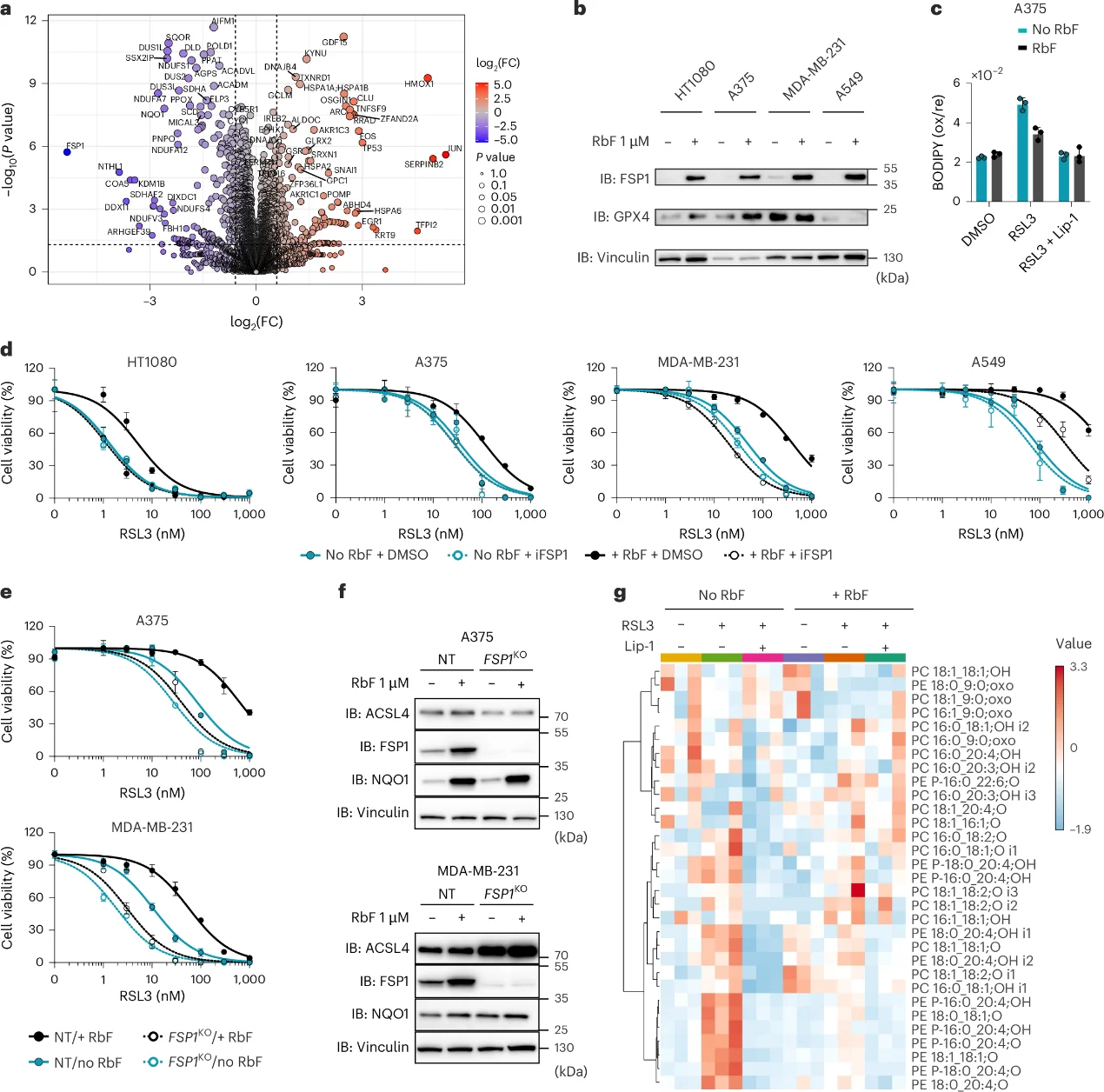
Fig. 3: Riboflavin availability as a central determinant of ferroptosis resistance
4. The Riboflavin Antimetabolite Roseoflavin Acts as a Potent Ferroptosis Sensitizer
In GPX4 KO/ FSP1 OE HT1080 cells, roseoflavin treatment induced cell death at nanomolar concentrations, which was entirely reversed by Lip-1 co-treatment. Crucially, roseoflavin-mediated sensitization to GPX4 inhibitors occurred selectively in cells possessing intact FSP1 expression, underscoring its precise target specificity for the FSP1 pathway.
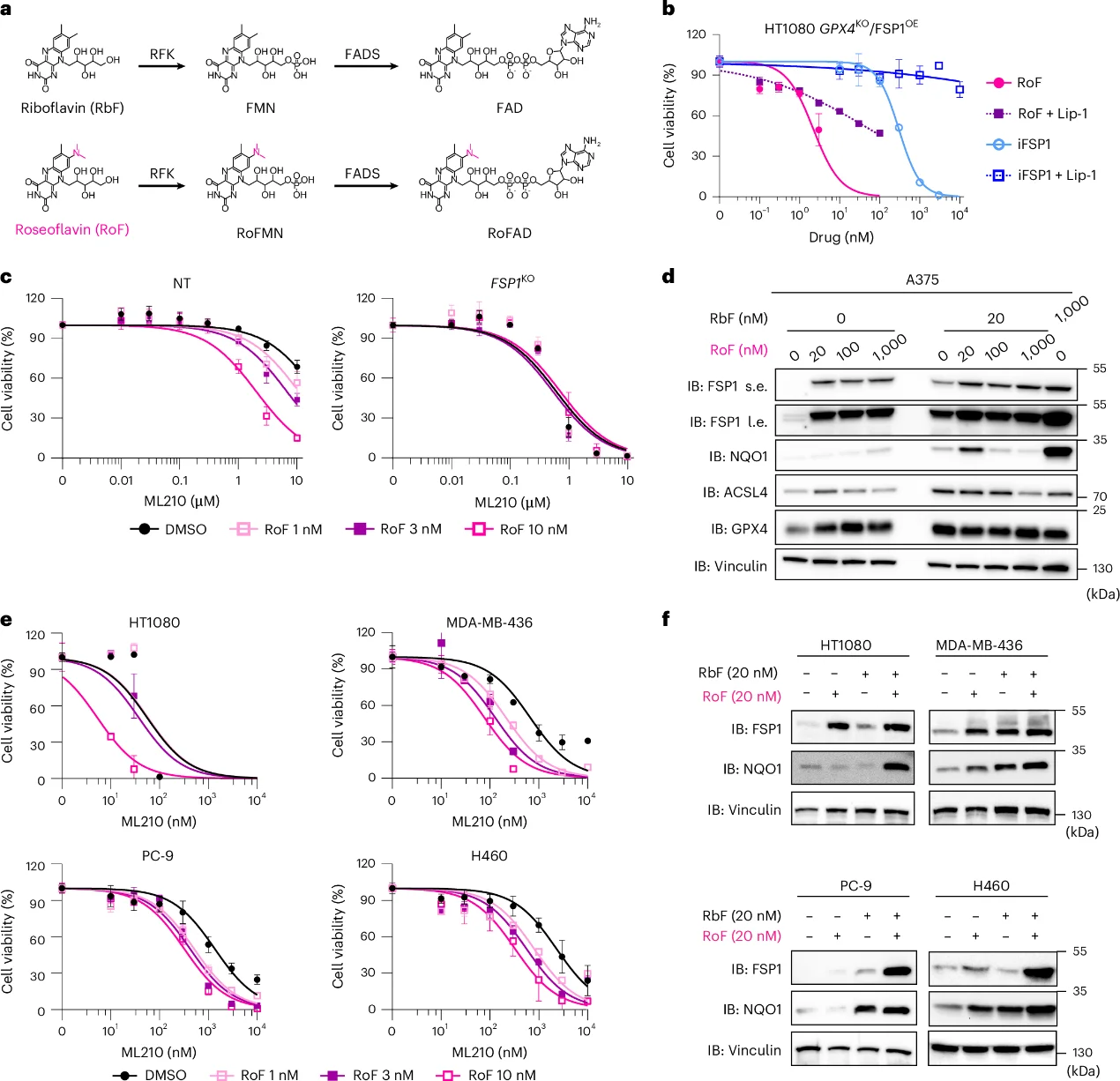
Fig. 4: The riboflavin antimetabolite roseoflavin promotes ferroptosis
5. Roseoflavin-Derived Metabolites Maintain FSP1 Structural Scaffolding but Abolish its Enzymatic Activity
To dissect the underlying biochemistry, FSP1-Flag complexes were purified via immunoprecipitation and subjected to a cell-free enzymatic activity assay. Exogenous roseoflavin supplementation failed to restore the metabolic reduction capacity of FSP1.
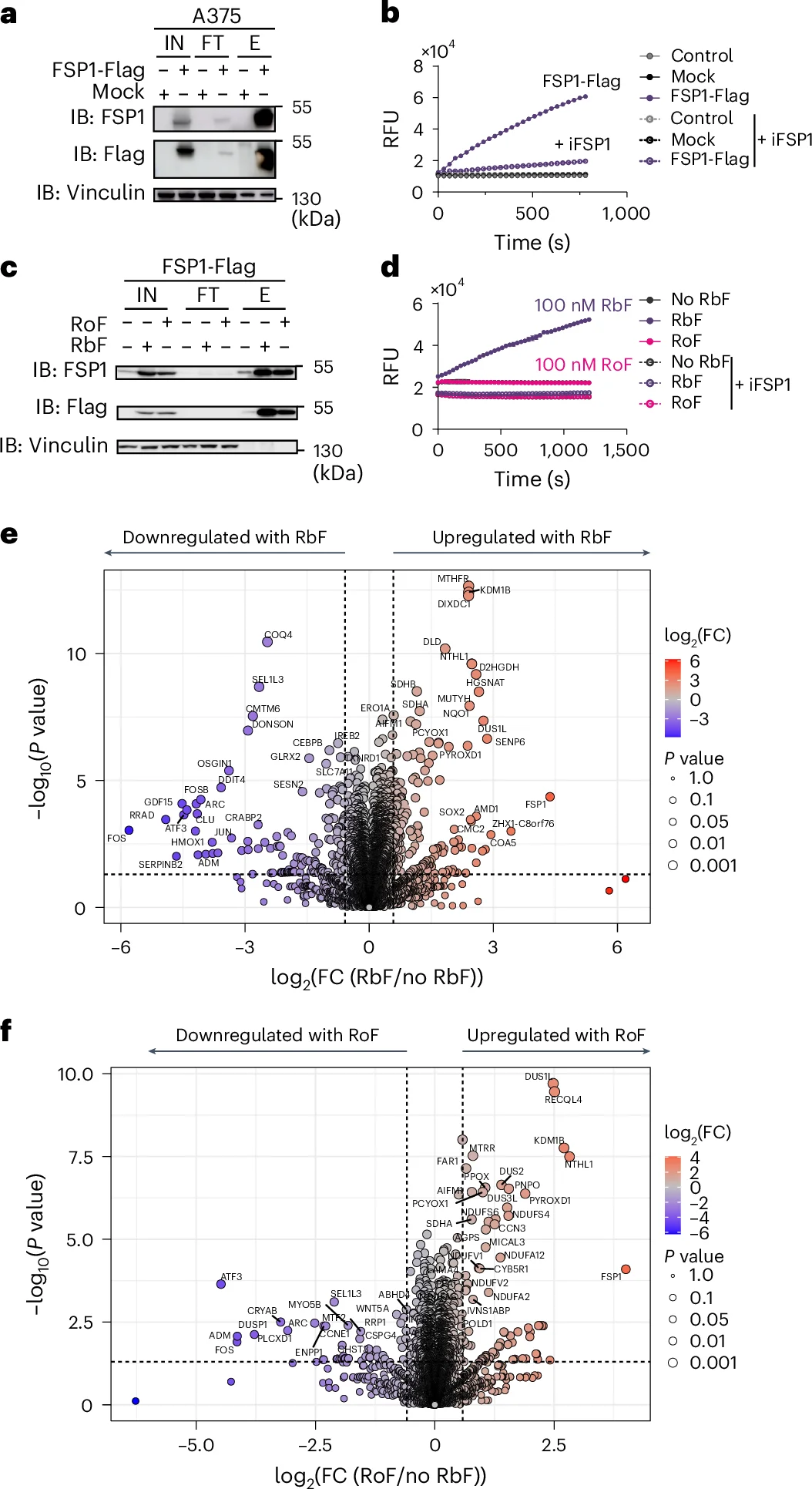
Fig. 5: The riboflavin antimetabolite roseoflavin disrupts FSP1 activity
6. Roseoflavin Binds FSP1 and Prevents Proteasomal Degradation
Structurally, roseoflavin differs from endogenous riboflavin via a dimethylamino substitution at the C8 position of the isoalloxazine ring. This modification severely alters the electron density distribution of the ring system. Consequently, the synthesized roseoflavin-adenine dinucleotide (roFAD) cofactor is incapable of accepting electrons from NAD(P)H, leaving FSP1 catalytically inert despite preserving its structural stability against proteasomal degradation.
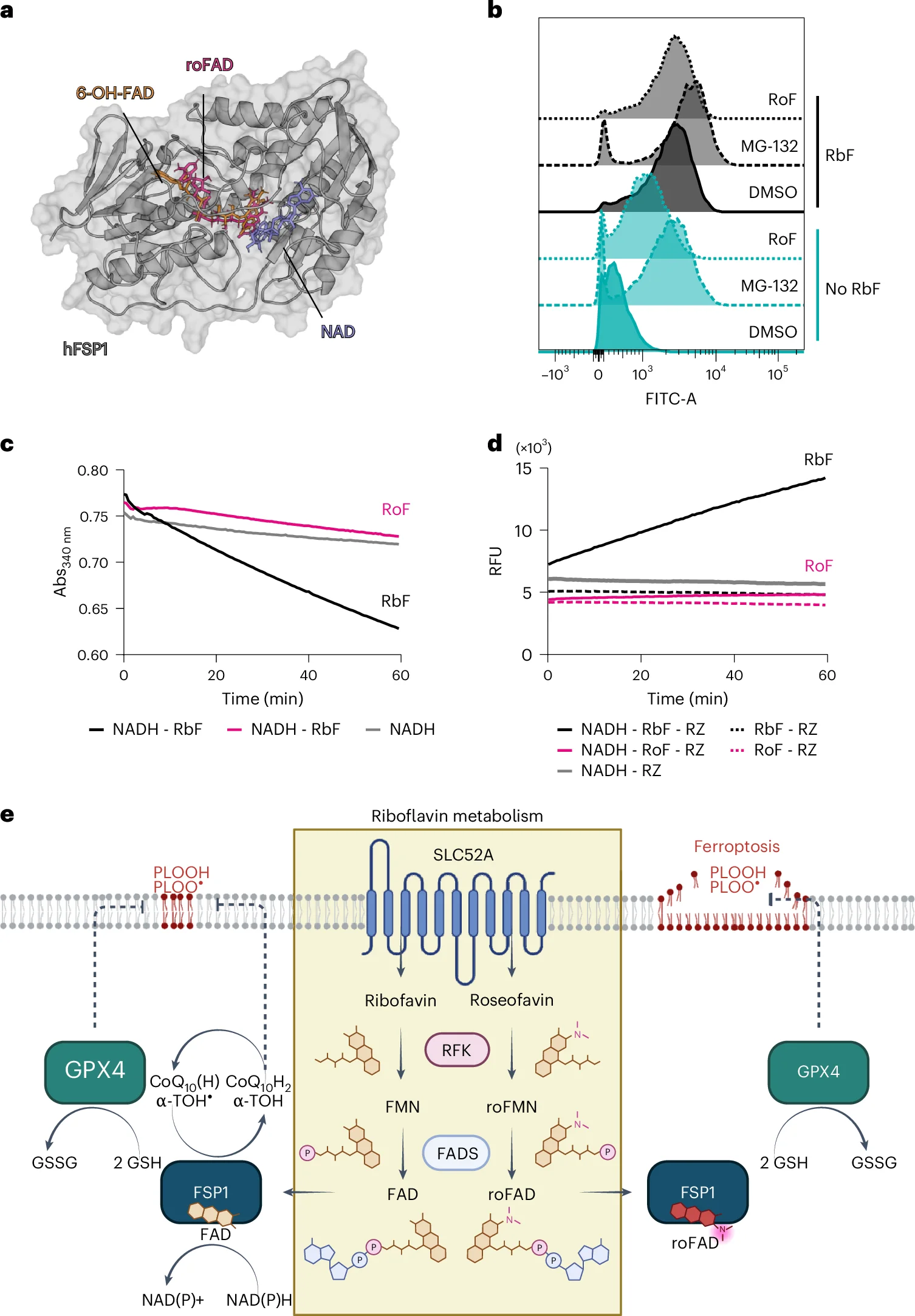
Fig. 6: Roseoflavin binds to FSP1 and prevents its degradation by the proteasome
Conclusion
This study defines RFK and FAD as critical upstream regulatory determinants of FSP1 stability. It demonstrates that metabolic flux through the precursor riboflavin pathway is essential for maintaining FSP1-mediated protection against ferroptotic lysis. Riboflavin deficiency not only impairs FSP1 protein homeostasis but drastically lowers the threshold for ferroptosis triggering. Beyond characterizing this endogenous metabolic requirement, the work highlights the therapeutic utility of riboflavin antimetabolites (e.g., roseoflavin) as chemical tools to disable FSP1 function. Collectively, these insights clarify how riboflavin metabolism fuels FSP1-driven lipid-soluble antioxidant cycling, offering vital implications for refining antioxidant-targeted clinical protocols.
CRISPR-iScreen™ Libraries from Ubigene
CRISPR-iScreen™ is an innovative technology independently developed by Ubigene. Currently, Ubigene offers over 40 types of off-the-shelf CRISPR libraries and one-stop CRISPR functional screening services (in vivo/in vitro) to support your research.
Contact us for more details >>> Promotions
Promotions 








