CT26.WT cell line, a magic route for colon cancer research & therapeutics
By Dr. ASAD UZZAMAN
Murine CT26 (Colon Tumor #26) cells were developed in 1995 by exposing BALB/c mice to N-nitroso-N-methylurethane (NMU) that form a rapid-growing grade IV carcinoma. These cells are easily implanted and readily metastasized. The cloned generated from CT26 cell line designated CT26.WT, adherent in nature and have a fibroblast-like morphology. The genome, transcriptome, and immunome studies revealed that CT26.WT cells possess a homozygous Kras mutation ( p.G12D), and a homozygous deletion (Cdkn2a). The proliferation and stem-cell markers (like Top2a, Birc5, Cldn6, and Mki67) are highly expressed while differentiation and top-crypt markers (like Muc2, Ms4a8a, and Epcam) are absent. CT26.WT cells share molecular features with aggressive, undifferentiated, refractory human colorectal carcinoma cells.
Colorectal cancer (CRC) is the third most common cancer in men and the second most common in women. Patients with stage IV CRC have a 5-year survival rate of 10–14%. Despite the technological advancements, survival for patients suffering from CRC has remained quite grim, with some biomarkers, such as the RAS mutation playing a major role in limiting the potential of therapeutic options. The pressure to improve the prognosis, therapeutics, and quality of life (QoL) for the patients suffering from this disease through the discovery of new diagnostic tools and therapeutic agents is ever increasing.
For the past few decades, monolayer cellular cultures, such as cancer cell lines or immortalized cell lines, have been widely applied in cancer research and these in vitro cultured cells are essential tools for the study of colorectal function, disease development, and therapeutic interventions. CT26.WT cell one of the popular cellular models as well as a tool for life scientists, who willing to study the mechanism of action of diseases or therapeutically active drug molecules as well as to decipher cell signaling events such as DNA damage repair. To date, CT26.WT cells have been described in more than 500 scientific publications.
Application:
CT26.WT has a wide application in the Bio-medical research arena.
1. BCancer vaccine: Study on genetically modified tumor cells for therapeutic cancer vaccine or preventive cancer vaccine.
2. Gene discovery: Identify new cancer driver genes and uncover cancer-specific vulnerabilities.
3. Cancer recurrence: Identify the cancer recurrence-associated genes, interaction network, downstream studies, and preventive measures of tumor recurrence.
4. New drugs: Novel therapeutic vulnerabilities and the development of more effective treatments.
5. Disease modeling: Allograft and xenograft modeling.
6. Phytomedicine: Effect of plant extract on tumor growth, metastasis, cytotoxicity, and apoptosis.
7. Oncolytic studies: Virus information, presence of genetic modifications and/or transgene expression, targeted cancer type, strategies, and safety profile.
CRISPR-U™ gene editing in CT26.WT cells
Genome editing is an addition, deletion, or replacement of a gene for wiping out or initiating explicit and preferred characters in the genome. The CRISPR system is used for precise genome editing and can generate gene knockout or knock-in genome manipulations through the substitution of a target genetic sequence with a desired donor sequence. CT26.WT cell is an aggressive murine colon cancer cell line. Gene editing within this cell line possible to generate single or multiple gene knockouts, mutation corrections, or insert reporter transgenes. These genome-edited cell lines will allow investigators to study cancer hallmarks, disclosure of drug resistance mechanism, cancer therapeutics, cell death research, functional genomics, signaling pathways, drug discovery, drug response, and cell therapy.
CRISPR/Cas9 technology positively fuel the advancement of in vivo and in vitro gene editing in colon cancer and have an immense impact on molecular medicine. Ubigene developed CRISPR-U™ for gene manipulation of the CT26.WT cell line. Thereby, possible to achieve genome editing in CT26.WT cells with the utilization of the CRISPR/Cas9 system. Ubigene can customize the gene-editing in eukaryotic CT26.WT cells as well as can generate various genes modification in animal models.
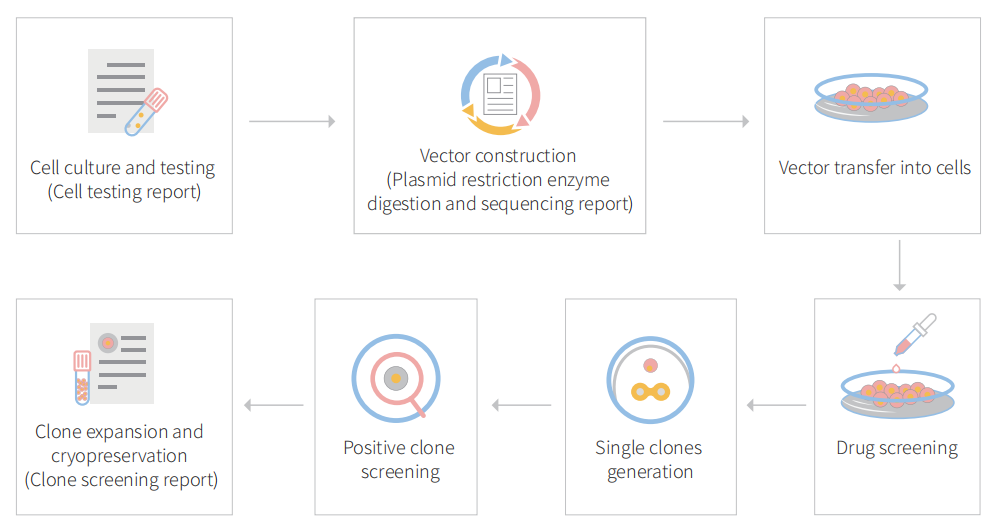
Figure: CRISPR-U™ customized workflow for engineered CT26.WT model cells
Case study :
Knockout cell line reveal that patients with immunoreactive tumors benefited from autophagy disruption
The role of autophagy in cancer is complex, context-dependent, and at times contradictory, as it has been shown to inhibit, promote, or be dispensable for tumor progression. The pro-survival functions of autophagy can be hijacked by cancer cells to enable rapid proliferation under conditions of metabolic stress, which are typically observed in the tumor microenvironment. In this study, the researcher evaluated the contribution of the immune system to the reliance of tumors on autophagy by depleting autophagy-related 7 (ATG7) in murine tumor cells and grafting them into immunocompetent versus immunodeficient hosts. They found that loss of ATG7 did not affect tumor growth in vitro or immunodeficient mice. Cancer cell reliance on autophagy was influenced by anti-tumor immune responses, including those mediated by CD8 + T cells.

Fig 1: Loss of ATG7 blocks autophagy and sensitizes cells to nutrient deprivation.
The researcher used the CRISPR/Cas9 strategy to knockout ATG7 from B16F10 (melanoma), MC38, and CT26 (colorectal carcinoma) cell lines to study the role of autophagy in murine cancer cells proliferation. Functional loss of ATG7 and autophagy were probed for ATG5, LC3, and p62 (Fig1a). All ATG7-deficient cell lines had a significant survival disadvantage (Fig1b-d). The proliferation of ATG7KO cell lines under nutrient-rich conditions showed that ATG7 did not impact the proliferation of B16F10, MC38, or CT26 cells in vitro (Fig1e-g).
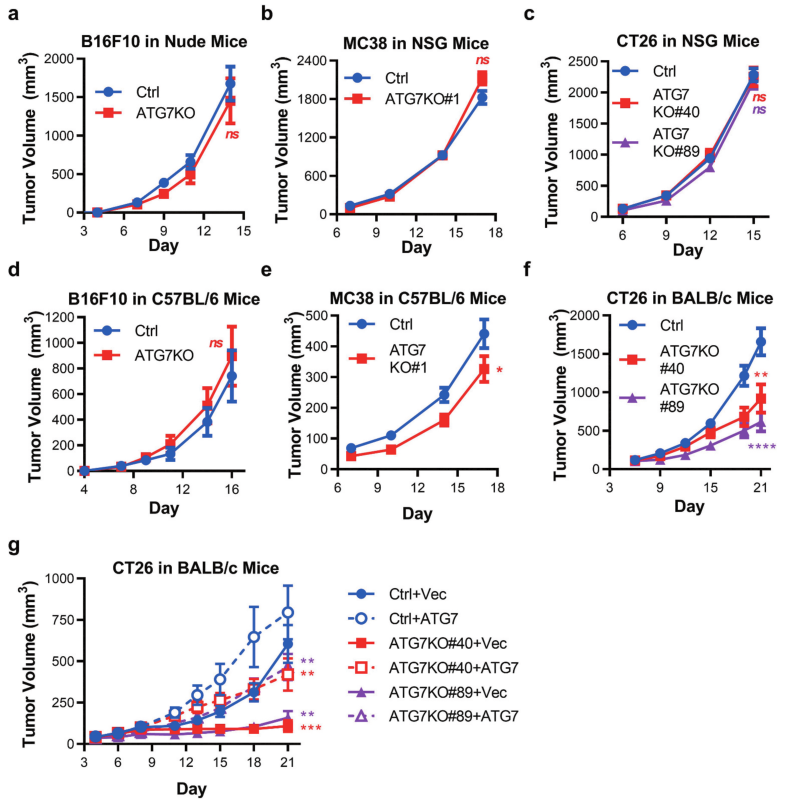
Fig 2: Murine tumors have differential reliance upon ATG7 when grown in immuno-competent hosts.
To determine autophagy can support murine cancer cell tumorigenicity in vivo, Ctrl, and ATG7KO cell lines were engrafted into immunodeficient mice. Loss of ATG7 did not impact the growth of B16F10 tumors (in nude mice), MC38, or CT26 tumors (in NSG mice) (Fig 2a-c). In vivo tumor growth of B16F10, MC38, or CT26 cell lines implanted in immune-competent (C57BL/6 or BALB/c) mouse strains shown in Fig 2d-f. In vivo tumor growth of CT26 tumor cells expressing vector control (+Vec) or ATG7 (+ATG7) implanted in BALB/c mice are shown in Fig 2g.
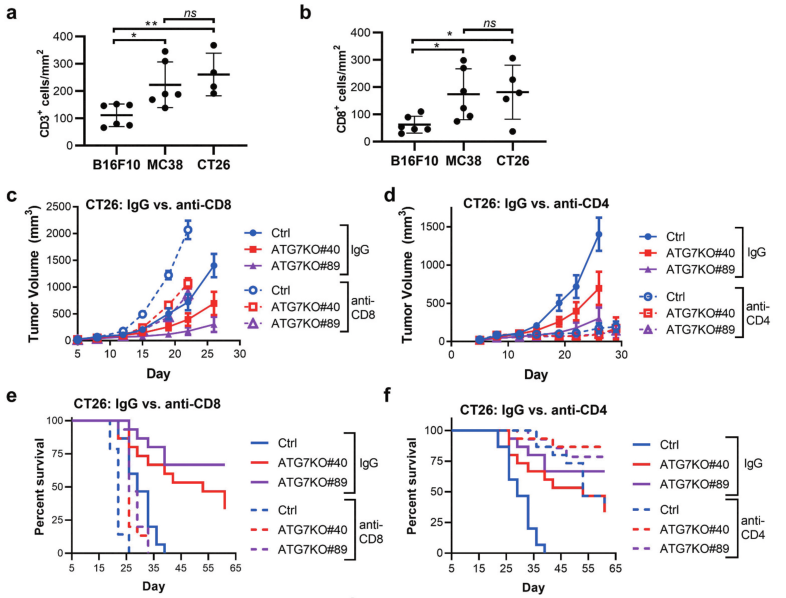
Fig 3: CD8 + and CD4 + T cell contribution to cancer-cell dependence on ATG7 in vivo.
To analyze levels of immune infiltrates in syngeneic models, the researcher applied immunohistochemistry (IHC) to detect and quantify CD3 + and CD8 + cells within B16F10, MC38, and CT26 tumors isolated from immunocompetent hosts (Fig 3a-b). Ctrl or ATG7KO CT26 cells implanted in BALB/c mice treated with IgG, anti-CD8, or anti-CD4 antibodies shown in Fig 3c-f.
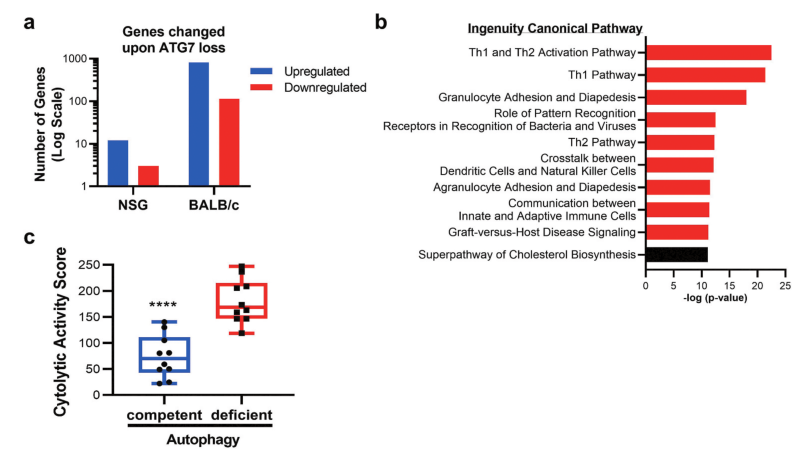
Fig 4: Gene expression and pathway alterations upon Atg7 loss.
Why autophagy disruption has more profound effects in the immunocompetent setting was performed on ATG7-competent or ATG7-deficient CT26 cells grown in immune-competent (BALB/c) or immune-deficient (NSG) mice. In NSG mice, loss of ATG7 in tumors resulted in the significant upregulation of 12 genes, and the significant downregulation of 3 genes when compared to autophagy-proficient tumors (Fig 4a). To decipher pathways impacted by autophagy disruption shown in Fig 4b. Autophagy-deficient tumors had an enhanced cytolytic activity score, as defined by log-average of Gzma and Prf1 expression (Fig 4c).
In summary, autophagy-deficient tumors had an abundance of cells with the potential to mediate tumor killing, such as cytotoxic CD8 + T cells or NK cells. The enrichment of immune-related genes indicates that disruption of cancer cell autophagy can enhance the recruitment, and possibly the activity, of effector cells to the tumor microenvironment, resulting in decreased tumor burden.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!

Reference:
Anti-tumor immunity influences cancer cell reliance upon ATG7. ONCOIMMUNOLOGY.2020, VOL. 9, NO. 1, e1800162.

 Subscribe Us
Subscribe Us Gene Editing Services
Gene Editing Services
 EZ-editor™
EZ-editor™ Red Cotton Gene knockout Project
Red Cotton Gene knockout Project





