Trust on CRISPR to stay younger

Aging is thought of as “the time-related deterioration of the physiological functions necessary for survival and fertility”. so, it is critical for scientists to find effective ways to study the molecular drivers of aging. Although, the molecular basis of physiological aging is adequately complex that researchers have not fully unraveled the processes and genes involved in it. Simply, to date the process of aging at the molecular level is not fully explored. There are nine traits that termed as potential hallmark of aging. Genomic instability, both internal and external factors that cause genetic damage can accelerate aging. Telomere attrition, telomeres protective “caps” getting shorter each time of cell division and over time, cells loses its dividing capability, which can lead to disease. Epigenetic alterations, changes in gene expression (not changes to the DNA itself) via an individual’s life experiences or environmental factors which affect aging. Loss of proteostasis, with age, cellular proteins become misfolded and therefore, lose their homeostatic functions (damaged proteins observation with aging or age-related diseases). Deregulated nutrient-sensing, especially metabolism-regulating pathways proteins (e.g. mTOR, sirtuins) are influenced by nutrient levels and promoting aging. Mitochondrial malfunction promotes aging. Altered intercellular communication malfunction resulting in inflammation and tissue damage. Cellular senescence impaired cell cleaning process, “Older” cells deposition can lead to harmful health effects. Finally, stem cell exhaustion reduces regenerating new tissue cells sponsoring aging.
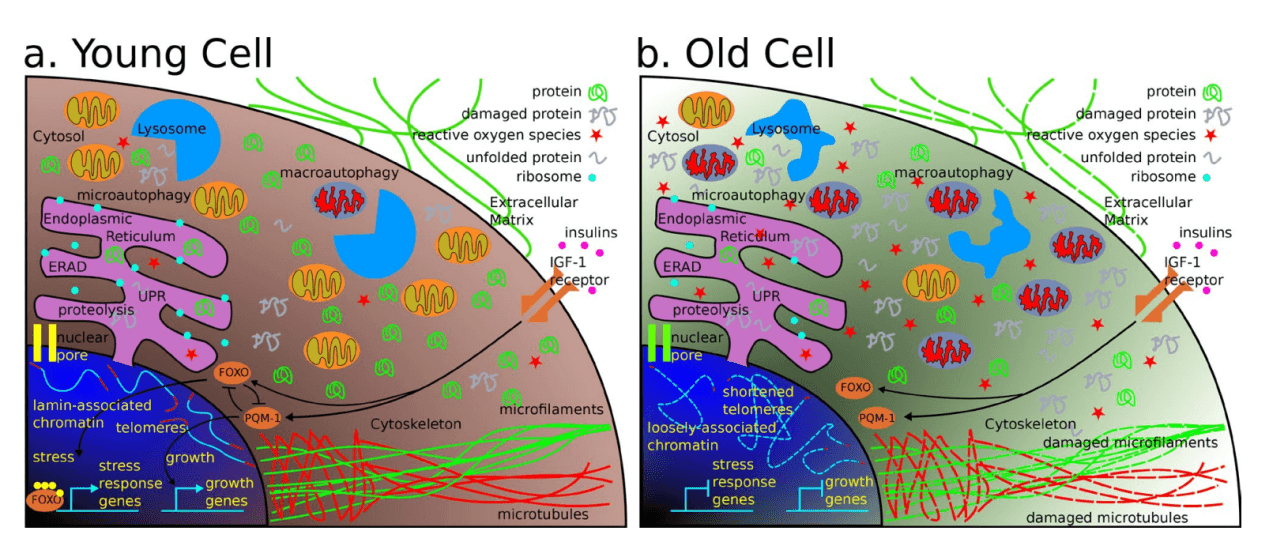
Aging is a leading risk factor for a number of debilitating conditions, including heart disease, cancer and Alzheimer's disease, to name a few. Although much remains to be discovered, patterns and connections are emerging, and this makes the needs of effective interventions for anti-aging therapies that targeting age-associated diseases. To date, researchers have found several genes in mammals that prolong lifespan, linking genetics to aging-related damage. If a gene is involved, it can be edited by CRISPR. However, editing a single gene in every cell type in the body is hard to do, if not impossible, in adults. Therefore, need to determine which cells could be edited, and which oxidative damage genes should be targeted? Similarly, instability of mitochondrial and genomic DNA are large contributors to age-related damage. This takes the form of small mutations in our DNA that occur and accumulate all over the body as cell types replenish and renew. In 2019, journal of Nature Medicine, highlight a novel CRISPR/Cas9 genome-editing therapy that can suppress the accelerated aging observed in mice with Hutchinson-Gilford progeria syndrome, a rare genetic disorder that also afflicts humans. This treatment provides important insight into the molecular pathways involved in accelerated aging, as well as how to reduce toxic proteins via gene therapy. Recent study suggests that cellular senescence is also a driver of aging and age-related pathology. Senescent cells are characterized not only by a cell-cycle arrest, but also by a potent pro-inflammatory phenotype, which is thought to drive aging and age-related diseases. Elimination of senescent cells in mice has been shown to reverse age-related pathology demonstrating that targeting senescent cells can be a powerful strategy to promote healthy aging.
CRISPR/Cas9 system can dramatically improve the physiological health and life span of human. Genome editing can provide a significant new understanding of how scientists may eventually be able to target molecular drivers of aging in humans. There will be limits to how far we can with such techniques. We could not reprogram the entire body, or enable people to live forever. But if we can repair some of the body’s worn out parts, that would allow us to enjoy a higher quality of life as we age. The newly developed CRISPR–Cas9 technology will not only support the development of genetic models of Aging & aging related diseases but also enhance our understanding of the pathogenesis of the related disorders including cancer, parkinson’s, huntington’s diseases, all of which have become more prevalent as the life expectancy of humans has increased. Novel genome editing technologies based on the CRISPR–Cas9 system together with powerful readout methods might help us better understand the logic of aging and unravel some of the mysteries of aging in general in the near future.
Application:
CT26.WT has a wide application in the Bio-medical research arena.
1. Genetics and functional genomics: mutations affecting longevity; gene homologies; organismal and cellular aging; gene manipulation.
2. Signaling and gene expression: mechanisms linking age-associated changes with phenotypes and physiology; intracellular signaling; interactions between cells and tissues; hormonal, immune, and inflammatory systems.
3. Cell proliferation, aging and death: replicative senescence; apoptosis; telomere biology and other intrinsic and extrinsic influences; chronological cellular aging; phenotypes of aging cells.
4. Cell stress and damage: extrinsic and intrinsic influences of free radicals on cells and tissues; free radical defense and damage; free radicals as signaling molecules; stress and aging.
5. Stem cells and aging: effects of age on stem cell generation; migration and homeostasis; stem cell-niche interactions; regulatory mechanisms.
6. Integrative physiology: outcomes of aging processes at cellular, molecular and organismal levels.
7. Potential clinical applications: leading to improved prevention, treatment of disease, a greater understanding of pathological processes with aging.
8. Drugs & Aging: drug discovery, screening, optimum drug therapy.
CRISPR-U™ gene editing for Aging research
According to WHO, at the biological level, aging results from the impact of the accumulation of a wide variety of molecular and cellular damage over time. There are nine traits that termed as potential hallmark of aging like genomic instability, telomere attrition, epigenetic alterations and loss of proteostasis etc. To date, aging researcher discovered several genes in mammals that prolong lifespan and linking genetics to aging-related damage. CRISPR-Cas system is a revolutionary genome-editing tool, which can use for precise genome editing and can generate gene knockout or knock-in genome manipulations through substitution of a target genetic sequence with a desired donor sequence. Gene-editing tools are capable of correcting gene-mediated age-related pathology, and therefore ameliorating or eliminating disease symptoms. Deleting target genes using the CRISPR system or correcting for gene mutations may ameliorate many different disorders detected in the aging population. Cancer cells targeted by the CRISPR system may result in an increased sensitivity to chemotherapeutics, lower proliferation, and higher cancer cell death. Ubigene developed CRISPR-U™ for gene manipulation of eukaryotic cell line. Thereby, possible to achieve genome editing in numerous eukaryotic cells with the utilization of the CRISPR/Cas9 system.
Case study :
CRISPR/Cas9 mediated study of mutation impact on telomere metabolism
Missense mutations in the human CTC1 gene cause Coats plus (CP), a rare autosomal recessive disorder characterized by retinal telangiectasia, intracranial calcifications, osteopenia, and gastrointestinal bleeding. Characterization of human disease mutations has often yielded valuable insights into basic biological functions. In this study researchers investigated the impact of CTC1L1142H on telomere metabolism and compared the effect of this CTC1 mutation in two distinct cell types, the HCT116 colon cancer cell line and the telomerase‐immortalized retinal pigment epithelial (RPE) cells. They found that mutant CTC1L1142H interacts poorly with STN1 and that the CTC1:STN1 subcomplex is sufficient to repress telomerase‐mediated telomere elongation. Expression of CP mutations that cannot interact with DNA Pol‐α show that CTC1:STN1 is also required to promote DNA Pol‐α‐mediated C‐strand maintenance. Thus, CST complex is required to coordinate both telomerase‐mediated G‐strand extension and DNA Pol‐α‐mediated C‐strand synthesis to maintain telomere length homeostasis.
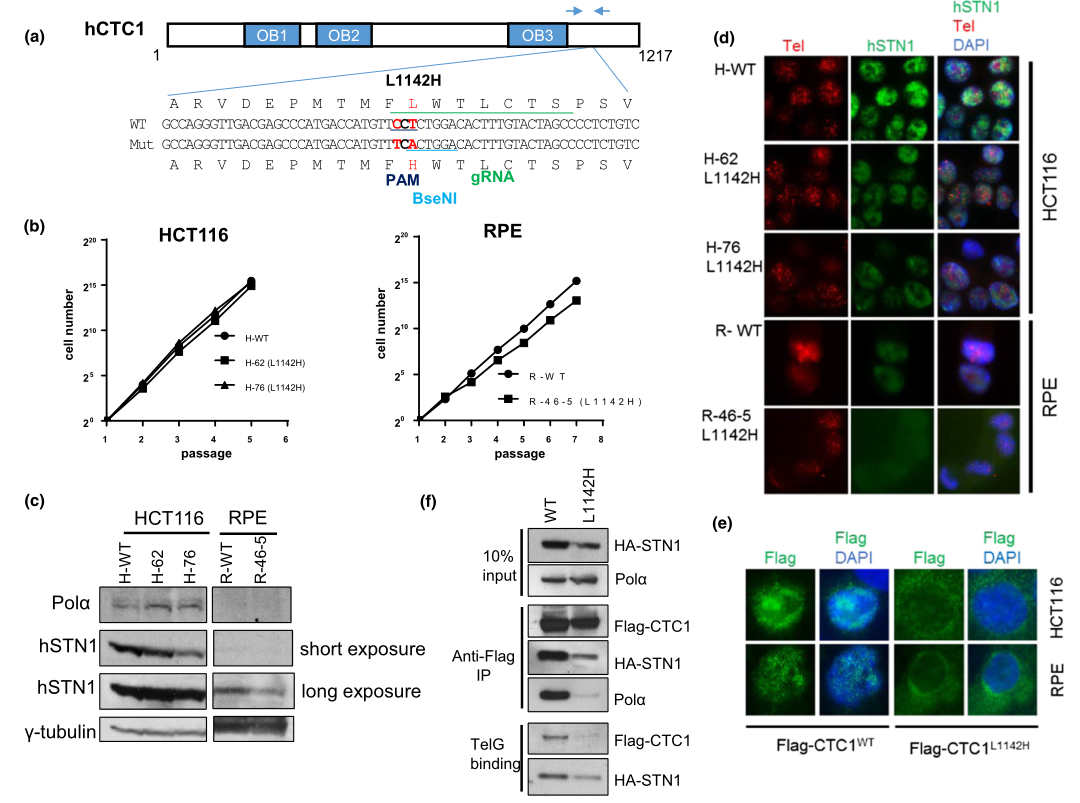
Fig 1: Generation of the CTC1 L1142H mutation in HCT116 and RPE cells using CRISPR/Cas9.
To understand mechanistically how the CTC1 L1142H mutation impacted telomere metabolism in CP patients, they utilized clustered, regularly interspaced, short palindromic repeats (CRISPR)/CRISPR-associated 9 (Cas9) to mutate CTC1 Leu 1142 to His 1142 on both alleles in the HCT116 cell line and telomerase-immortalized RPE cells. The mutant cell line were used to measure the proliferative capacities, expression pattern of endogenous DNA Pol-a and STN1, Immuno-FISH analysis for endogenous STN1 (green) and telomeres (red) in WT or L1142H mutant HCT116 or RPE cell lines. They found that CTC1, in complex with STN1, negatively regulates telomere length. While the CTC1 L1142H mutation led to an initial increase in telomere length in RPE cells, this increase in length cannot be stably maintained.
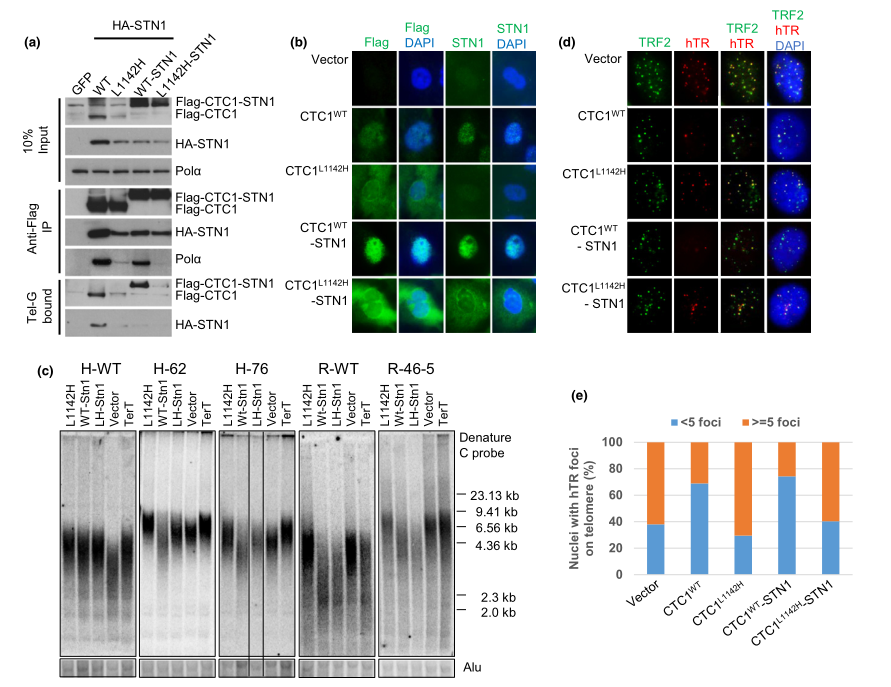
Fig 2:The CTC1:STN1 complex inhibits telomerase recruitment to telomeres.
The CTC1 L1142H mutant interacts poorly with both STN1 and DNA Pol-a and fails to bind ss telomeric DNA, suggesting that physical interactions between CTC1, STN1, and DNA Pol-a are all required to bind to ss telomeric DNA. To test this hypothesis, researcher examined whether artificially tethering mutant Flag-CTC1 L1142H to STN1 via a flexible 10-amino acid linker could rescue CTC1 L1142H ’s interaction with DNA Pol-a and ss telomeric DNA. The Flag-CTC1 WT -linker-STN1 protein interacted robustly with both DNA Pol-a and ss telomeric DNA (Figure 2a), completely localized to the nucleus (Figure 2b) and functionally reduced telomere lengths in both WT and CTC1 L1142H HCT116 and RPE cells (Figure 2c). In addition, expression of the Flag-CTC1 WT -linker-STN1 constructs reduced telomerase accumulation on telomeres, as revealed by telomerase FISH (Figure 2d and e). They found that CTC1 is required to directly interact with STN1 to form a CTC1:STN1 (C:S) complex. C:S then interacts with DNA Pol-a to enable stable binding to ss telomeric DNA, and this C:S: DNA Pol-a complex is inhibitory to telomerase-mediated G-strand extension.
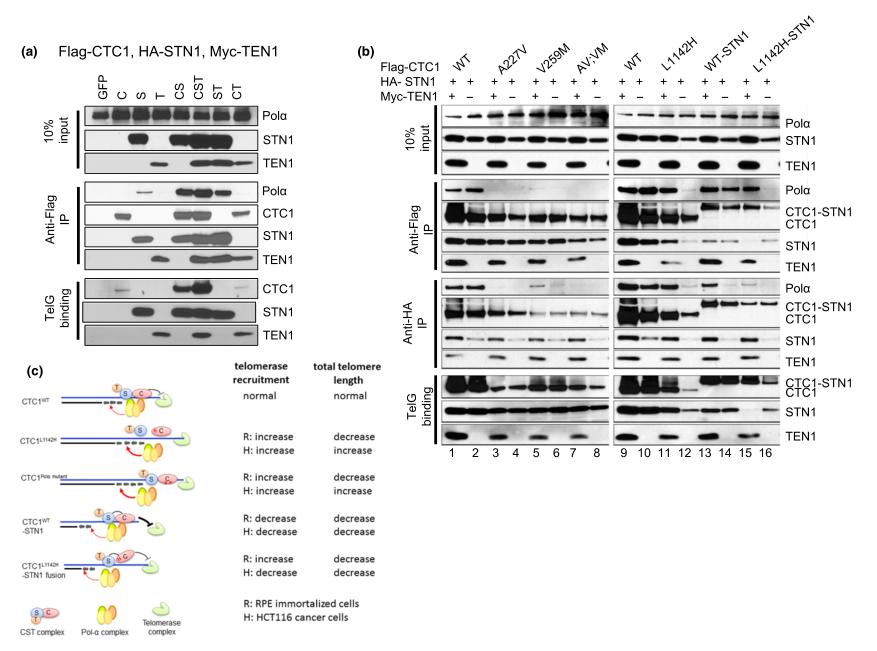
Fig 5:TEN1 enhances CTC1:STN1 interaction.
To examine the contribution of TEN1 to CST complex formation with DNA Pol-a and ss telomeric DNA, they first expressed Flag-CTC1, HA-STN1, and Myc-TEN1 in HEK293T cells and examined their interactions by Co-IP. By itself, HA-STN1, but not Flag-CTC1 or Myc-TEN1, weakly interacted with endogenous DNA Pol-a (Figure 3a). Co-expressing all three C:S:T components together resulted in robust binding to DNA Pol-a, although C:S and S:T also interacted well with DNA Pol-a. The presence of Myc-TEN1 enhanced the interaction between Flag-CTC1 L1142H and HA-STN1, as well as complex formation between Flag-CTC1 L1142H , HA-STN1, and DNA Pol-a(Figure 3b). Thus, provide functional evidence that CTC1:STN1 is required to repress telomerase activity in vivo. The CTC1 L1142H protein interacts poorly with STN1 and localizes partially to the cytoplasm, leading to telomerase-mediated telomere elongation (Figure 2c).
In summary, the CTC1 L1142H :STN1:TEN1 complex cannot compete with telomerase for access to the 3 prime G-rich overhang. Impaired interaction between CTC1 L1142H :STN1 and DNA Pol-a results in increased telomerase recruitment to telomeres and telomere elongation, further revealing that C:S binding to DNA Pol-a is required to fully repress telomerase activity. In addition, C:S regulates C-strand fill-in by DNA Pol-a. These findings confirm the CST complex as the major regulator of both G-strand extension and C-strand fill-in reactions.
Ubigene developed CRISPR-U™ which optimizes eukaryotic cells and animal gene-editing vectors and processes. The efficiency and accuracy are 10x higher than traditional methods. Contact us immediately to know about your research related services!

Reference:
CTC1-STN1 coordinates G- and C-strand synthesis to regulate telomere length. Aging Cell, 2018 ;17(4):e12783.

 Subscribe Us
Subscribe Us Gene Editing Services
Gene Editing Services
 EZ-editor™
EZ-editor™ Red Cotton Gene knockout Project
Red Cotton Gene knockout Project





