How can CRISPR facilitates Alzheimer's disease research?
At present, there are at least 50 million dementia patients in the world, which is expected to reach 152 million by 2050, of which about 60%-70% are patients with Alzheimer's disease. Alzheimer's disease (AD) is a central neurodegenerative disease characterized by cognitive decline and memory impairment.
In recent years, CRISPR/Cas9 gene-editing technology has developed rapidly, showing great potential in basic scientific research and the development of human disease treatment targets. For a long time, CRISPR has been evaluated as a valuable tool for the research of AD. It is mainly used in AD model construction, pathogenic gene screening and targeted therapy. Up to now, many studies have focused on in-depth research on AD by CRISPR. Here, we have selected some researches on AD using gene editing technology in recent years.
CRISPR/Cas9 editing of APP C-terminus attenuates β-cleavage and promotes α-cleavage(Nature communications,IF=14.9196(2021))
CRISPR/Cas9 guided gene-editing is a potential therapeutic tool, however current approaches relying on canonical gene-deletion or mutation-correction using CRISPR-technology are limited in practicability and scope. First, elimination of entire genes would almost certainly have deleterious effects on physiology, since most of these genes have normal roles as well. Secondly, strategies aimed at correcting point-mutations would only be applicable to the small fraction of neurodegenerative diseases that are inherited (typically < 10% of cases). Based on this, Krishanu Saha from the University of Wisconsin cooperated with Subhojit Roy to use CRISPR/Cas9 technology to edit the C-terminal of amyloid precursor protein (APP) to attenuating APP-β cleavage and Aβ production in AD. It provides a strategy to favorably manipulate the amyloid pathway to intervene the process of AD by gene editing.
In this study, they introduced a CRISPR/Cas9-based strategy in cell and animal models to edit endogenous amyloid precursor protein (APP) at the extreme C-terminus and reciprocally manipulate the amyloid pathway. It was found that this editing changed the cleavage balance of APP in human IPSC derived neuronal cells, i.e. attenuating APP-β-cleavage and Aβ production, while up-regulating neuroprotective APP-α-cleavage. Further, during the analysis of the possible off target effect of gene editing, it was found that APP N-terminus and compensatory APP-homologues remain intact, with no apparent effects on neurophysiology in vitro. And robust APP-editing is seen in human iPSC-derived neurons and mouse brains with no detectable off-target effects. Finally, in order to explain the regulation mechanism of APP β/α by this gene editing, they used AAV9s for in vivo injection into mouse hippocampus. They further prove that this strategy likely works by limiting APP and BACE-1 approximation, and they also delineate mechanistic events that abrogates APP/BACE-1 convergence in this setting.

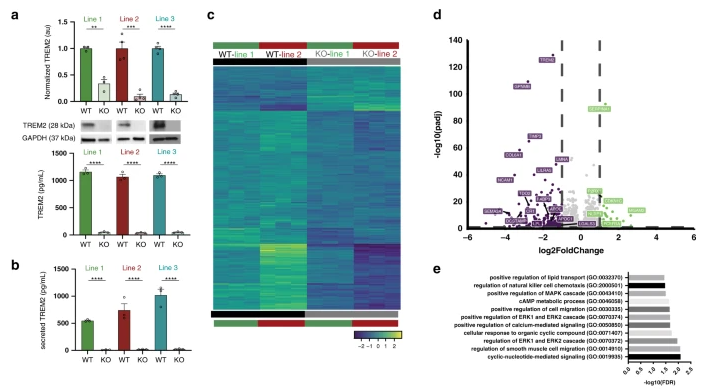
Gene expression and functional deficits underlie TREM2-knockout microglia responses in human models of Alzheimer's disease(Nature communications,IF=14.9196(2020))
The discovery of TREM2 as a myeloid-specific Alzheimer’s disease (AD) risk gene has accelerated research into the role of microglia in AD. While TREM2 mouse models have provided critical insight, the normal and disease-associated functions of TREM2 in human microglia remain unclear. In order to further explore the impact of TREM2 on human diseases, Mathew blurton Jones’ team from the University of California Irvine studied the function of TREM2 in human microglia, and published the research results in the journal Nature communications.
They profile microglia differentiated from isogenic, CRISPR-modified TREM2-knockout induced pluripotent stem cell (iPSC) lines. By combining transcriptomic and functional analyses with a chimeric AD mouse model, they find that TREM2 deletion reduces microglial survival, impairs phagocytosis of key substrates including APOE, and inhibits SDF-1α/CXCR4-mediated chemotaxis, culminating in an impaired response to beta-amyloid plaques in vivo. In addition, single-cell sequencing of xenotransplanted human microglia further highlights a loss of disease-associated microglial (DAM) responses in human TREM2 knockout microglia that we validate by flow cytometry and immunohistochemistry. Taken together, these studies reveal both conserved and novel aspects of human TREM2 biology that likely play critical roles in the development and progression of AD.
Ubigene’s KO cell bank offers TREM2 knockout cell line. As low as $1980, deliver in 1 week. Click here to explore your KO cell lines>>
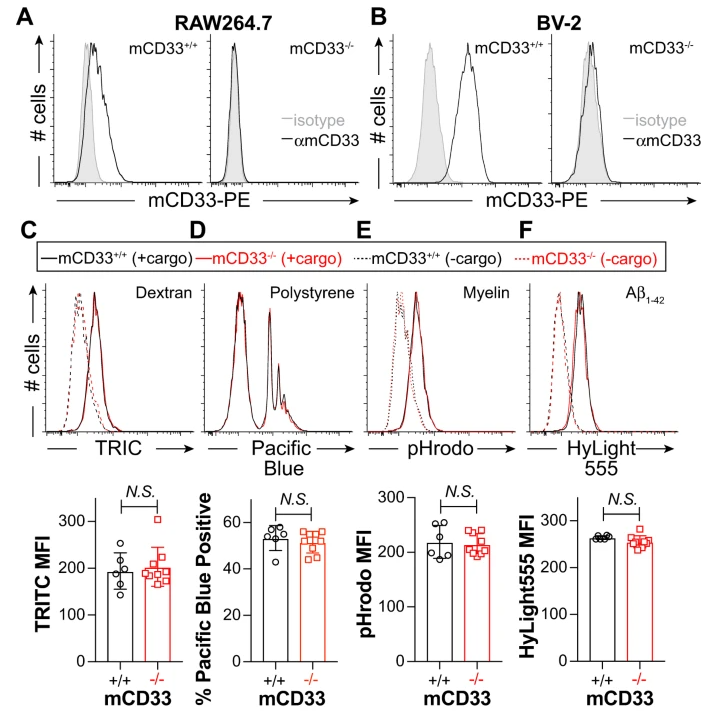
Repression of phagocytosis by human CD33 is not conserved with mouse CD33(Communications biology,IF= 6.268(2019))
CD33 is an immunomodulatory receptor linked to Alzheimer’s disease (AD) susceptibility via regulation of phagocytosis in microglia. Divergent features between human CD33 (hCD33) and murine CD33 (mCD33) include a unique transmembrane lysine in mCD33 and cytoplasmic tyrosine in hCD33. The functional consequences of these differences in restraining phagocytosis remains poorly understood. In a research report published in the international journal Communications Biology, scientists from the University of Alberta in Canada discovered that mCD33 and hCD33 have divergent roles in regulating phagocytosis, highlighting the importance of studying hCD33 in AD susceptibility. In addition, they also developed a new transgenic mouse model expressing hCD33 in microglia, which provides a suitable model for exploring the relationship between hCD33 and amyloid plaque accumulation.
Using a new αmCD33 monoclonal antibody, we show that mCD33 is expressed at high levels on neutrophils and low levels on microglia. Notably, cell surface expression of mCD33 is entirely dependent on Dap12 due to an interaction with the transmembrane lysine in mCD33. In RAW264.7 cultured macrophages, BV-2 cultured microglia, primary neonatal and adult microglia, uptake of cargo — including aggregated Aβ1-42 — is not altered upon genetic ablation of mCD33. Alternatively, deletion of hCD33 in monocytic cell lines increased cargo uptake. Furthermore, transgenic mice expressing hCD33 in the microglial cell lineage showed repressed cargo uptake in primary microglia. Therefore, mCD33 and hCD33 have divergent roles in regulating phagocytosis, highlighting the importance of studying hCD33 in AD susceptibility.
Ubigene has successfully edited thousands of genes in about 200 cell lines, and successfully constructed a Knockout Cell Line Bank with nearly 2000 genes. In-stock KO cell line starting from $1980, deliver in 1 week! In addition, Ubigene also provides gene knock-in and point mutation cell model construction services to meet various research needs.

 Subscribe Us
Subscribe Us Gene Editing Services
Gene Editing Services
 EZ-editor™
EZ-editor™ Red Cotton Gene knockout Project
Red Cotton Gene knockout Project





