Exclusive CRISPR-U™ technology and stem cell culture system
facilitate iPSC gene editing
In August, Ubigene introduced the relevant
CRISPR/Cas9 gene editing applications of iPSC in disease modeling, drug screening and gene therapy (Figure
1). Those who have not our previous atricle please click here >>What
happened when iPSC met CRISPR?). After the article released, we received many inquiries, such as cell culturing,
single-cell cloning, low positive rate, etc...To facilitate your iPSC gene editing experiments, we sorted
out the issues that may happened and the relevant solutions. Hope this article could
help!

Figure.1 Clinical applications of iPSC gene
editing
Issues of iPSC
culturing
1)Poor cell viability
during culturing and easy to differentiate
Unlike tumor cells, iPSCs are more delicate. Here are some tricks to maintain cell
viability and stemness of iPSCs. The basic aspect is the selection of culture
medium and matrix. High-quality reagents must be
selected to ensure adequate nutrition and a comfortable growth environment. The culture system used after thawing is recommended to be
consistent with that before freezing. If the replacement of culture medium or matrix is necessary, it can only be replaced during
passage, and cells would need some time
to adapt. Moreover, unlike other tumor cells that can refresh culture medium once every two
or three days, iPSCs need to refresh culture medium every day and subculture in time to avoid affecting cell activity and causing differentiation due
to overgrowth or contact inhibition of colonies. During subculture, it should also be noted that cells
cannot be over digested. In addition, the culture of iPSCs needs
constant attention and care,
including but not limited to observing iPSCs under phase contrast microscope (4x, 10x, 20x and 40x
magnification) every day, and monitoring iPSC colony morphology, differentiation and confluence (Figure 2). The ideal colony
of iPSC morphology should be compact inside, uniform in
size and clear in edge. If there are a few differentiated cells at the edge, timely and
adequately subculture should be made to maintain the
pluripotency and logarithmic growth of iPSC.
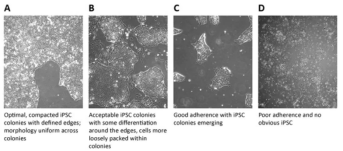
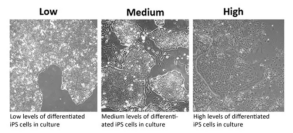
Figure 2. iPSC colony ranking and differentiation levels (Source:
sigmaaldrich)
2)Difficult to isolate
single-cell clones
Colony
growth and expansion is the key stage to obtain positive gene-editing cells. This process is time-consuming
and laborious. If the rate of colony formation is low, the workload will increase. The cell condition before
cloning is very important. The better the cell condition is, the higher the rate of colony formation is. In
addition, iPSC is easy to apoptosis after being digested into a single cell, and apoptosis inhibitor must be
added to make the clones grow better.
Low gene editing
efficiency in iPSCs
1)Strategy
design and transfection efficiency
The targeting
strategy design is crucial to the
success of the iPSC gene-editing project. For the knockout
project, the selection of knockout size, the specificity and the cutting efficiency of sgRNA could affect the chance of getting positive
cells. For
point mutation or knockin project, the distance between the
cutting
position and the mutation locus
or knockin
position should be
comprehensively considered. Meanwhile, the introduction of synonymous
mutation at PAM position and the design of the donor homologous
arm should also be taken into account. A good editing strategy is the basis for the
successful implementation of the project. In addition, iPSCs from different individuals and
tissues are quite different, and the transfection methods and parameters may be different. It is necessary to
conduct preliminary experiments for each
iPSC, to optimize
the transfection parameters and improve the
transfection rate.
The improvement of transfection can greatly increase the success of gene editing.
2)HDR
efficiency
Compared with gene knockout, the project difficulty of point
mutation and knockin in iPSC is
higher. The main reason is that compared with immortalized
cell lines, the efficiency of directed homologous recombination repair (HDR) in iPSCs is low [1]. HDR is the main method to repair
CRISPR induced double strand breaks (DSB) by exogenous
donor DNA. In
order to overcome the low HDR efficiency, researchers have adopted several strategies, such as adding resistance
genes to CRISPR plasmids or donor DNA. Although this method is effective, it will insert foreign DNA into the
genome [2]; Using lox franked selection markers or PiggyBAC
transposon system can improve HDR efficiency, but its construction cycle is also significantly prolonged [3]; Although the method of
single-strand oligonucleotide (ssODN) donor
avoids the problem of random integration of large double strand DNA molecules, it still suffers from the
problems of low success rate of repair and sequence disorder around DSB sites [4]; There are also
methods such as
using known cell cycle inhibitors to synchronize cell cycle to achieve the purpose of timing delivery of Cas9
RNP complex [5], and using small
molecule compounds
to inhibit non homologous end direct connection (NHEJ). Among them, the addition of NHEJ inhibitor is more
convenient and efficient, which is a common improvement method.
3)Genotyping
As
mentioned earlier, the workload of iPSC colony isolation is large. Before isolating single cells, strict
quality control should be carried out on the transfected cell pool to ensure that the editing on the cell
pool level is effective. Ubigene’s genotype analysis system (GAS) can be used for
analyzing transfected cell pool genotype, confirming editing efficiency, guiding whether to proceed next
step and the number of single cells should be isolated, achieving high-throughput interpretation of
monoclonals genotype, screening positive clones, and saving a lot of labor. Click here
to get a free trial>>
Case study
Using RNP method to achieve c.G2149T point mutation in iPSC. After transfection, the
recombination efficiency of the
cell pool was detected. According to the Sanger sequencing result, gRNA had
significant cutting efficiency (Figure. 3). According to the
EZ-editorTM GAS, homologous recombination
genotype accounted for 14% (Figure. 4), which was high in
efficiency. Single-cell clone
isolation could be performed.
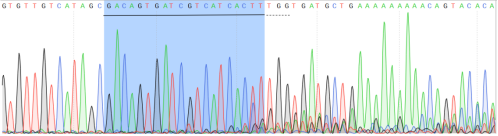
Figure 3.
Sanger sequencing result of transfected cell pool

Figure 4. EZ editorTM GAS result of HDR analysis
The issues and solutions are sorted out for you. You could now practice with
confidence!
If you still hesitate to do it
yourself, Ubigene provides customized services for
iPSC knockout, knockin and point
mutation. Our experience in iPSC gene editing ensures the
delivery of high-quality gene-edited iPSCs. We have optimized stem cell media to easily improve iPSCs culture, tested at least three transfection methods for different iPSCs to ensure transfection efficiency, and invented unique EZ-editorTM monoclone validation technology to high-throughput screen for positive
clones. Click
to learn more details
of iPSC gene
editing services>>
Related
products:
Monoclone Validation
Kit>>Allow early colony
validation during monoclonal growth. 30-5 cells can get enough templates for PCR, only 15 minutes, amplify
sequence as long as 10kb!
References:
[1] Hockemeyer D,
Jaenisch R. Induced
pluripotent stem cells meet genome editing[J]. Cell stem cell, 2016, 18(5): 573-586.
[2] Zhang Y, Schmid B,
Nielsen T T, et
al. Generation of a human induced pluripotent stem cell line via CRISPR-Cas9 mediated integration of a
site-specific homozygous mutation in CHMP2B[J]. Stem cell research, 2016, 17(1): 151-153.
[3] Yusa K, Rashid S
T, Strick-Marchand
H, et al. Targeted gene correction of α1-antitrypsin deficiency in induced pluripotent stem cells[J]. Nature,
2011, 478(7369): 391-394.
[4] Yang L, Guell M,
Byrne S, et al.
Optimization of scarless human stem cell genome editing[J]. Nucleic acids research, 2013, 41(19):
9049-9061.
[5] Lin S, Staahl B T,
Alla R K, et al.
Enhanced homology-directed human genome engineering by controlled timing of CRISPR/Cas9 delivery[J]. elife,
2014, 3: e04766.

 Subscribe Us
Subscribe Us Gene Editing Services
Gene Editing Services
 EZ-editor™
EZ-editor™ Red Cotton Gene knockout Project
Red Cotton Gene knockout Project





